organic papers
o2164
Hempelet al. C15H14N4S doi:10.1107/S1600536805018453 Acta Cryst.(2005). E61, o2164–o2166 Acta Crystallographica Section E
Structure Reports Online
ISSN 1600-5368
2-(Biphenyl-2-yl)-5-(1-methylhydrazino)-1,3,4-thiadiazole: a thiadiazole–hydrazine
derivative with anticonvulsant properties
Andrew Hempel,aLilian Y. Y. Ma,aNorman Camerman,a,b* Donald Mastropaoloband Arthur Camermanb†
aDepartment of Biochemistry, University of
Toronto, Medical Sciences Building, Toronto, Canada M5S 1A8, andbArdono Research, 341
101st Avenue SE, Bellevue, WA 98004, USA
† Deceased.
Correspondence e-mail: norman.camerman@utoronto.ca
Key indicators Single-crystal X-ray study
T= 294 K
Mean(C–C) = 0.009 A˚
Rfactor = 0.071
wRfactor = 0.136
Data-to-parameter ratio = 11.7
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2005 International Union of Crystallography Printed in Great Britain – all rights reserved
In the title compound, C15H14N4S, the outer phenyl ring makes
an angle of 101.4 (2) with the plane through the inner benzene ring, and the planes of the thiadiazole ring and the attached benzene ring intersect at an angle of 146.7 (2). In addition to weak N—H N hydrogen bonds, creating a two-dimensional network parallel to the bc plane of the crystal structure, there is also one non-standard hydrogen-bond interaction of the type C—H N. Stereochemical comparison with the closely related compound 1-[5-(biphenyl-2-yl)-1,3,4-thiadiazol-2-yl]methanaminium chloride shows that the two compounds utilize the same mechanism for anticonvulsant activity.
Comment
Promising anticonvulsant activity has been reported in some members of a series of synthetic substituted thiadiazole hydrazines (Chapleo et al., 1986). Subsequently, aminoalkyl analogues were prepared in order to avoid the presence of a potentially troublesome free hydrazine group (Stillingset al., 1986), and a number of those derivatives were equally potent anticonvulsants. We determined the structure of the most potent of the aminoalkyl thiadiazole hydrazine derivatives, 1-[5-(biphenyl-2-yl)-1,3,4-thiadiazol-2-yl]methanaminium chloride, (II), and despite its chemical dissimilarity to conventional antiepileptic drugs such as phenytoin, we were able to identify stereochemical features in common which may be responsible for their similar anticonvulsant properties (Camermanet al., 2005). We now report the structure of the most potent of the original hydrazine series, 2-biphenyl-2-yl-5-(1-methylhydrazino)-1,3,4-thiadiazole, (I), and show that it too contains the stereochemical properties correlated with anticonvulsant activity.
The structure of (I) is presented in Fig. 1. Bond distances and angles are within normal ranges, even with higher than usual values for the anisotropic displacement parameters, especially for the atoms of the outer phenyl ring. The sum of the angles at the amino atom N7 is 327, indicating sp3 hybridization. The torsion angles N3—C2—N6—N7 and N3— C2—N6—C8 are 173.6 (5) and 15.9 (7), respectively. The
thiadiazole ring is planar. The plane of the outer phenyl ring makes an angle of 101.4 (2) with that of the inner benzene ring, which in turn intersects the plane of the thiadiazole ring at an angle of 146.7 (2). For comparison, the same parameters in (II) are 98.3 (2) and 165.5 (4), respectively.
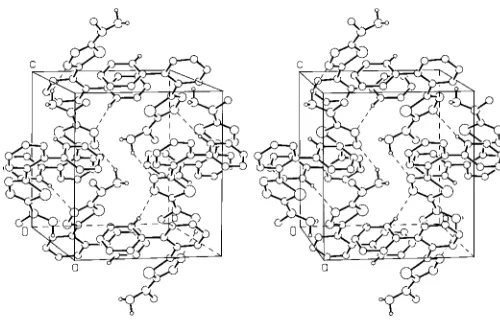
Weak N—H N hydrogen bonds (Table 1) produce a two-dimensional network of molecules parallel to the bc plane (Fig. 2), and create distinct hydrophobic (benzene rings) and hydrophilic (thiadiazole ring) parallel regions. A non-standard C—H N interaction and van der Waals contacts also contribute to the crystal packing. The weak intermolecular interactions between the molecules and the existence of an empty 40.2 A˚3volume in the hydrophobic environment of the
benzene rings contribute substantially to the loose crystal packing and the high thermal displacement parameters of the benzene rings.
We have superimposed the structures of (I) and the aminoalkyl derivative, (II); the results are shown in Fig. 3. The overall stereochemistry and the spatial arrangement of the
electronegative atoms is very similar in the two active compounds. It is extremely likely, therefore, that the two compounds exert their anticonvulsant properties through the same mechanism.
Experimental
The title compound was obtained from Reckitt & Colman plc, UK. Extensive crystallization experiments to find proper crystallization conditions produced only crystals of low quality. The crystal used was obtained by slow evaporation of a 1:1 petroleum ether–ethyl acetate solution at 294 K. Efforts to obtain better crystals were unsuccessful.
Crystal data
C15H14N4S
Mr= 282.36 Monoclinic, P21=c a= 13.886 (3) A˚ b= 9.898 (2) A˚ c= 10.896 (2) A˚
= 91.46 (2) V= 1497.1 (5) A˚3
Z= 4
Dx= 1.253 Mg m
3
CuKradiation Cell parameters from 32
reflections
= 19–38
= 1.88 mm1 T= 294 (2) K Needle, colourless 0.250.070.04 mm
Data collection
Picker FACS-1 four-circle diffractometer
!/2scans
Absorption correction: scan (Northet al., 1968) Tmin= 0.851,Tmax= 0.926
2259 measured reflections 2259 independent reflections
1111 reflections withI> 2(I)
max= 62.5
h= 0!15 k= 0!11 l=9!12 3 standard reflections
every 100 reflections intensity decay: 2.3%
Refinement
Refinement onF2 R[F2> 2(F2)] = 0.071
wR(F2) = 0.136
S= 0.89 2259 reflections 193 parameters
H atoms treated by a mixture of independent and constrained refinement
w= 1/[2
(Fo2) + (0.0477P)2]
whereP= (Fo2+ 2Fc2)/3
(/)max< 0.001
max= 0.28 e A˚
3
min=0.17 e A˚
3
Extinction correction:SHELXL97 (Sheldrick, 1997)
Extinction coefficient: 0.0014 (3)
organic papers
Acta Cryst.(2005). E61, o2164–o2166 Hempelet al. C
[image:2.610.44.298.75.247.2]15H14N4S
o2165
Figure 1
The molecular structure of (I), showing 35% probability displacement ellipsoids. H atoms are drawn as small circles of arbitrary radii.
Figure 2
[image:2.610.316.554.75.227.2]A stereoscopic view of the molecular packing and hydrogen-bond scheme (shown as dashed lines between atoms). Atoms are drawn as circles of arbitrary radii. For clarity, only H atoms involved in the hydrogen bonding are shown.
Figure 3
[image:2.610.44.294.532.697.2]Table 1
Hydrogen-bond geometry (A˚ ,).
D—H A D—H H A D A D—H A
N7—H71 N4i
0.99 (6) 2.21 (7) 3.118 (7) 151 (5) N7—H72 N3ii 0.91 (6) 2.20 (7) 3.100 (7) 169 (5) C16—H16 N3iii
0.93 2.62 3.534 (8) 168
Symmetry codes: (i)x;yþ1 2;zþ
1
2; (ii)xþ1;y 1 2;zþ
3
2; (iii)x;yþ 1 2;z
1 2.
All H atoms were visible in a difference map. However, due to the paucity of intensity data, the H atoms were refined using a riding-model approximation, except for the two H atoms from the amino group, which were taken from the difference map and refined freely. One overall displacement parameter was refined for H atoms in the methyl group [Uiso(H) = 0.143 (15) A˚
2] and another for the remaining
benzene-ring H atoms [0.123 (7) A˚2]. The range of C—H distances is 0.93–0.96 A˚ and the range of N—H distances is 0.91–0.99 (6) A˚
Data collection: Picker Operating Manual (Picker, 1967); cell refinement:Picker Operating Manual; data reduction:DATRDNin
The XRAY System(Stewart, 1976); program(s) used to solve struc-ture:SHELXS97(Sheldrick, 1997); program(s) used to refine struc-ture: SHELXL97(Sheldrick, 1997); molecular graphics: ORTEP-3 for Windows(Farrugia, 1997); software used to prepare material for publication:SHELXL97.
References
Camerman, A., Hempel, A., Mastropaolo, D. & Camerman, N. (2005).Acta Cryst.C61, o427–o430.
Chapleo, B. C., Myers, M., Myers, P. M., Saville, J. F., Smith, A. C. B., Tulloch, I. F., Walter, D. S. & Welbourn, A. P. (1986).J. Med. Chem.29, 2273– 2280.
Farrugia, L. J. (1997).J. Appl. Cryst.30, 565.
North, A. C. T., Phillips, D. C. & Mathews, F. S. (1968).Acta Cryst.A24, 351– 359.
Picker (1967).Picker Operating Manual. Picker, Cleveland, Ohio, USA. Sheldrick, G. M. (1997). SHELXS97andSHELX97. University of Go¨ttingen,
Germany.
Stewart, J. M. (1976).DATRDNin The XRAY System of Crystallographic Programs. Editor. Technical Report TR-446. Computer Science Center, University of Maryland, College Park, Maryland, USA.
Stillings, M. R., Welbourn, A. P. & Walter, D. S. (1986).J. Med. Chem.29, 2280–2284.
organic papers
o2166
Hempelet al. Csupporting information
sup-1 Acta Cryst. (2005). E61, o2164–o2166
supporting information
Acta Cryst. (2005). E61, o2164–o2166 [https://doi.org/10.1107/S1600536805018453]
2-(Biphenyl-2-yl)-5-(1-methylhydrazino)-1,3,4-thiadiazole: a thiadiazole
–
hydrazine derivative with anticonvulsant properties
Andrew Hempel, Lilian Y. Y. Ma, Norman Camerman, Donald Mastropaolo and Arthur
Camerman
2-(Biphenyl-2-yl)-5-(1-methylhydrazino)-1,3,4-thiadiazole
Crystal data
C15H14N4S
Mr = 282.36
Monoclinic, P21/c
a = 13.886 (3) Å
b = 9.898 (2) Å
c = 10.896 (2) Å
β = 91.46 (2)°
V = 1497.1 (5) Å3
Z = 4
F(000) = 592
Dx = 1.253 Mg m−3
Cu Kα radiation, λ = 1.54178 Å
Cell parameters from 32 reflections
θ = 19–38°
µ = 1.88 mm−1
T = 294 K
Needle, colorless 0.25 × 0.07 × 0.04 mm
Data collection
Picker FACS-1 four-circle diffractometer
Radiation source: fine-focus sealed tube Ni filtered radiation monochromator
θ/2θ scans
Absorption correction: ψ scan
(North et al., 1968)
Tmin = 0.851, Tmax = 0.926 2259 measured reflections
2259 independent reflections 1111 reflections with I > 2σ(I)
Rint = 0.000
θmax = 62.5°, θmin = 3.2°
h = 0→15
k = 0→11
l = −9→12
3 standard reflections every 100 reflections intensity decay: 2.3%
Refinement
Refinement on F2
Least-squares matrix: full
R[F2 > 2σ(F2)] = 0.071
wR(F2) = 0.136
S = 0.89
2259 reflections 193 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H atoms treated by a mixture of independent and constrained refinement
w = 1/[σ2(F
o2) + (0.0477P)2] where P = (Fo2 + 2Fc2)/3 (Δ/σ)max < 0.001
Δρmax = 0.28 e Å−3 Δρmin = −0.17 e Å−3
supporting information
sup-2 Acta Cryst. (2005). E61, o2164–o2166
Special details
Experimental. Generally no observable reflections above θ = 60° due to poor quality of the crystal and high anisotropic
temperature displacement parameters. A few visible reflections from the region θ = 60–65° have been identified on the
Weissenberg photographs and duly collected.
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used
only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2
are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
S1 0.29751 (10) 0.10959 (11) 0.65738 (10) 0.0663 (5)
C2 0.3943 (3) 0.1980 (4) 0.7163 (4) 0.0554 (12)
N3 0.4067 (3) 0.3187 (4) 0.6690 (3) 0.0672 (11)
N4 0.3356 (3) 0.3456 (4) 0.5818 (3) 0.0659 (11)
C5 0.2746 (3) 0.2488 (4) 0.5631 (4) 0.0546 (12)
N6 0.4544 (3) 0.1436 (4) 0.8035 (3) 0.0712 (12)
N7 0.4229 (4) 0.0191 (4) 0.8514 (4) 0.0691 (13)
C8 0.5231 (4) 0.2243 (6) 0.8746 (5) 0.0957 (19)
H8A 0.5403 0.3028 0.8283 0.143 (15)*
H8B 0.5798 0.1716 0.8925 0.143 (15)*
H8C 0.4947 0.2518 0.9500 0.143 (15)*
C9 0.1910 (4) 0.2645 (4) 0.4771 (4) 0.0654 (14)
C10 0.1511 (4) 0.1594 (5) 0.4071 (4) 0.0695 (14)
C11 0.0748 (4) 0.1868 (6) 0.3240 (5) 0.100 (2)
H11 0.0471 0.1168 0.2783 0.123 (7)*
C12 0.0408 (5) 0.3162 (7) 0.3097 (6) 0.125 (3)
H12 −0.0080 0.3339 0.2519 0.123 (7)*
C13 0.0781 (5) 0.4192 (6) 0.3795 (6) 0.123 (2)
H13 0.0527 0.5058 0.3716 0.123 (7)*
C14 0.1534 (4) 0.3947 (5) 0.4618 (5) 0.0907 (17)
H14 0.1795 0.4656 0.5076 0.123 (7)*
C15 0.1850 (4) 0.0177 (5) 0.4161 (5) 0.0659 (14)
C16 0.2673 (6) −0.0234 (8) 0.3623 (6) 0.126 (3)
H16 0.3051 0.0392 0.3218 0.123 (7)*
C17 0.2961 (7) −0.1603 (10) 0.3672 (7) 0.154 (4)
H17 0.3505 −0.1886 0.3264 0.123 (7)*
C18 0.2457 (7) −0.2468 (8) 0.4293 (9) 0.141 (3)
H18 0.2657 −0.3363 0.4344 0.123 (7)*
C19 0.1665 (6) −0.2092 (7) 0.4852 (9) 0.147 (4)
H19 0.1305 −0.2724 0.5274 0.123 (7)*
C20 0.1374 (4) −0.0762 (6) 0.4807 (6) 0.107 (2)
H20 0.0833 −0.0503 0.5235 0.123 (7)*
supporting information
sup-3 Acta Cryst. (2005). E61, o2164–o2166
H72 0.477 (5) −0.033 (7) 0.854 (5) 0.13 (3)*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
S1 0.0930 (10) 0.0403 (6) 0.0643 (8) −0.0036 (7) −0.0231 (6) 0.0088 (6)
C2 0.061 (3) 0.048 (3) 0.057 (3) 0.004 (2) −0.010 (2) −0.001 (2)
N3 0.085 (3) 0.052 (2) 0.065 (3) −0.014 (2) −0.008 (2) 0.000 (2)
N4 0.093 (3) 0.048 (2) 0.055 (2) −0.010 (2) −0.017 (2) 0.0016 (18)
C5 0.076 (3) 0.042 (3) 0.045 (3) 0.002 (2) −0.006 (3) 0.003 (2)
N6 0.089 (3) 0.061 (3) 0.063 (3) −0.002 (2) −0.024 (2) 0.000 (2)
N7 0.095 (4) 0.051 (3) 0.060 (3) 0.008 (2) −0.018 (3) 0.005 (2)
C8 0.114 (5) 0.101 (4) 0.071 (4) −0.023 (4) −0.023 (4) 0.006 (3)
C9 0.088 (4) 0.039 (3) 0.068 (3) −0.005 (2) −0.015 (3) 0.008 (2)
C10 0.098 (4) 0.048 (3) 0.062 (3) −0.004 (3) −0.017 (3) 0.012 (2)
C11 0.120 (5) 0.067 (4) 0.109 (5) −0.009 (4) −0.058 (4) 0.008 (3)
C12 0.136 (6) 0.083 (4) 0.153 (6) −0.005 (4) −0.081 (5) 0.031 (4)
C13 0.139 (6) 0.067 (4) 0.160 (6) 0.005 (4) −0.062 (5) 0.021 (4)
C14 0.111 (5) 0.049 (3) 0.111 (4) 0.007 (3) −0.030 (4) 0.006 (3)
C15 0.073 (4) 0.059 (3) 0.066 (3) −0.013 (3) −0.010 (3) −0.002 (3)
C16 0.178 (8) 0.102 (6) 0.099 (5) 0.007 (5) 0.045 (5) 0.022 (4)
C17 0.205 (10) 0.128 (7) 0.131 (7) 0.063 (7) 0.073 (6) −0.013 (6)
C18 0.162 (9) 0.060 (4) 0.200 (10) 0.018 (6) −0.019 (8) −0.015 (5)
C19 0.112 (6) 0.062 (5) 0.268 (11) −0.007 (4) 0.011 (7) 0.042 (5)
C20 0.082 (5) 0.065 (4) 0.174 (6) −0.002 (3) 0.008 (4) 0.018 (4)
Geometric parameters (Å, º)
S1—C2 1.715 (4) C11—C12 1.373 (8)
S1—C5 1.743 (4) C11—H11 0.9300
C2—N3 1.314 (5) C12—C13 1.366 (8)
C2—N6 1.361 (5) C12—H12 0.9300
N3—N4 1.378 (5) C13—C14 1.382 (7)
N4—C5 1.292 (5) C13—H13 0.9300
C5—C9 1.482 (6) C14—H14 0.9300
N6—N7 1.412 (5) C15—C20 1.349 (7)
N6—C8 1.453 (6) C15—C16 1.360 (8)
N7—H71 0.99 (6) C16—C17 1.413 (10)
N7—H72 0.91 (6) C16—H16 0.9300
C8—H8A 0.9600 C17—C18 1.306 (10)
C8—H8B 0.9600 C17—H17 0.9300
C8—H8C 0.9600 C18—C19 1.324 (11)
C9—C14 1.399 (6) C18—H18 0.9300
C9—C10 1.396 (6) C19—C20 1.377 (8)
C10—C11 1.402 (6) C19—H19 0.9300
C10—C15 1.482 (7) C20—H20 0.9300
supporting information
sup-4 Acta Cryst. (2005). E61, o2164–o2166
N3—C2—N6 123.4 (4) C13—C12—C11 120.6 (5)
N3—C2—S1 115.2 (3) C13—C12—H12 119.7
N6—C2—S1 121.4 (3) C11—C12—H12 119.7
C2—N3—N4 110.3 (3) C12—C13—C14 120.0 (5)
C5—N4—N3 114.9 (3) C12—C13—H13 120.0
N4—C5—C9 121.5 (4) C14—C13—H13 120.0
N4—C5—S1 112.6 (3) C13—C14—C9 120.9 (5)
C9—C5—S1 125.7 (3) C13—C14—H14 119.6
C2—N6—N7 114.4 (4) C9—C14—H14 119.6
C2—N6—C8 122.5 (4) C20—C15—C16 116.5 (6)
N7—N6—C8 119.2 (4) C20—C15—C10 121.8 (6)
N6—N7—H71 112 (4) C16—C15—C10 121.7 (6)
N6—N7—H72 104 (4) C17—C16—C15 120.7 (7)
H71—N7—H72 111 (5) C17—C16—H16 119.7
N6—C8—H8A 109.5 C15—C16—H16 119.7
N6—C8—H8B 109.5 C18—C17—C16 119.6 (8)
H8A—C8—H8B 109.5 C18—C17—H17 120.2
N6—C8—H8C 109.5 C16—C17—H17 120.2
H8A—C8—H8C 109.5 C17—C18—C19 121.1 (8)
H8B—C8—H8C 109.5 C17—C18—H18 119.5
C14—C9—C10 118.7 (4) C19—C18—H18 119.5
C14—C9—C5 117.1 (4) C18—C19—C20 119.9 (8)
C10—C9—C5 124.1 (4) C18—C19—H19 120.0
C11—C10—C9 119.4 (4) C20—C19—H19 120.0
C11—C10—C15 117.3 (4) C15—C20—C19 122.0 (7)
C9—C10—C15 123.2 (4) C15—C20—H20 119.0
C10—C11—C12 120.4 (5) C19—C20—H20 119.0
C10—C11—H11 119.8
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
N7—H71···N4i 0.99 (6) 2.21 (7) 3.118 (7) 151 (5)
N7—H72···N3ii 0.91 (6) 2.20 (7) 3.100 (7) 169 (5)
C16—H16···N3iii 0.93 2.62 3.534 (8) 168