Acta Cryst.(2001). E57, o99±o101 DOI: 101107/S1600536800020079 Alex F. T. Yokochiet al. C15H22O
o99
organic papers
Acta Crystallographica Section E Structure Reports Online
ISSN 1600-5368
Vulgarone B
Alex F. T. Yokochi,a* James D.
Whiteaand George Sturtzb
aDepartment of Chemistry, Oregon State
University, Corvallis, Oregon, USA, and bAromaGen, Albany, Oregon, USA
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study T= 293 K
Mean(C±C) = 0.003 AÊ Disorder in main residue Rfactor = 0.050 wRfactor = 0.132
Data-to-parameter ratio = 10.9
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2001 International Union of Crystallography Printed in Great Britain ± all rights reserved
The crystal structure of vulgarone B (2,6,6,11-tetramethyltri-cyclo[5.4.0.02,8]undec-10-en-9-one), a carbocyclic
sesquiter-pene with the formula C15H22O, is reported. All
intramolecular geometric parameters are as expected. The molecule contains a four-membered ring, in which all atoms are stereogenic. The 1,3 and 2,4 atoms of this cyclobutane are bridgehead C atoms which form part of six-membered and seven-membered rings.
Comment
Continuing our structural and synthetic studies of the effects of ring strain on the properties of carbocyclic natural products (White & Lee, 1997), we report here the structure of vulgarone B, (I). This sesquiterpene was isolated as the major terpenoid component (ca 40%) of the essential oil of Arte-misia douglasianaBess., a plant that occurs widely in Western Oregon. An extract of this plant has been found to possess potent insecticidal and gastropod repellent activity. Previously, vulgarone B was isolated along with its isomer vulgarone A as a minor constituent of the oil of the medicinal plant
Chry-santhemum vulgare (L.) Bernh, [Tanacetum vulgare (L)]
(Uchioet al., 1977). It has also been obtained from the volatile oil of cultivars ofSantolina chamaecyparcissusL. (Baiget al., 1989).
The structure of vulgarone B, including its absolute con®guration, had earlier been deduced from spectroscopic evidence and by chemical correlation with the related sesquiterpene (+)--longipinene (Uchio, 1978). The results from our X-ray single-crystal diffraction experiment con®rm the assigned structure. We also attempted to determine the absolute con®guration of vulgarone B from the diffraction data, but, due to the small magnitude of the anomalous scat-tering components, the re®ned value of the absolute structure parameter could not be reliably determined. This is re¯ected in the fact that the model shown in Fig. 1 yields a value of
ÿ0.7 (6) for this parameter, whereas the inverted model yields a value of 1.7 (6). Further, since the absolute structure derived
organic papers
o100
Alex F. T. Yokochiet al. C15H22O Acta Cryst.(2001). E57, o99±o101from the model shown is identical to the previously deduced con®guration for this molecule (Uchio, 1978), we ®nd it reasonable to conclude that this is the true con®guration of vulgarone B.
All the observed intramolecular distances and angles are well within the expected values. The most interesting feature of the molecule is the four-membered ring, in which all four atoms are stereogenic. Opposite corners of this ring are the bridgehead C atoms that are components of two other rings,
i.e.a seven-membered cycle and a membered ring. The six-membered ring contains a C CÐC O moiety, which should force ®ve of the six atoms in the ring to be planar. This is found to be correct; atoms O1, C1, C2, C3, C31, C4 and C11 are nearly coplanar, with a maximum deviation from the mean plane of 0.028 (2) AÊ by atom C2. This geometric constraint also causes a slight distortion of the four-membered ring, in which the bridgehead C atoms of the six-membered ring have moved toward each other. This is re¯ected in the CÐCÐC angles within the ring, which are identical pairwise at about 84 and 89as shown in Table 1.
Experimental
Crystals of the title compound were obtained by sublimation of a bulk sample under ambient pressure and temperature. Due to the high vapor pressure of the compound, it also sublimes during the diffraction experiment. Thus, in order to minimize the rate of subli-mation, the crystal was completely encapsulated in a thin layer of epoxy glue. This strategy was suf®cient to allow data collection over a period of a few days.
Crystal data
C15H22O
Mr= 218.33
Orthorhombic,P212121
a= 6.628 (1) AÊ
b= 10.218 (1) AÊ
c= 19.889 (1) AÊ
V= 1347.0 (3) AÊ3
Z= 4
Dx= 1.077 Mg mÿ3
CuKradiation Cell parameters from 99
re¯ections = 8.0±27.3
= 0.50 mmÿ1
T= 293 (2) K Block, colorless 0.40.40.3 mm
Data collection
SiemensP4 diffractometer !/2scans
Absorption correction: empirical
via scans (Northet al., 1968) usingXEMP(Siemens, 1990)
Tmin= 0.827,Tmax= 0.866
2270 measured re¯ections 1911 independent re¯ections 1666 re¯ections withI> 2(I)
Rint= 0.041
max= 67.6
h=ÿ6!7
k=ÿ12!12
l=ÿ23!23 3 standard re¯ections
every 97 re¯ections intensity decay: 1%
Re®nement
Re®nement onF2
R(F) = 0.050
wR(F2) = 0.132
S= 1.07 1911 re¯ections 176 parameters
H-atom parameters constrained
w= 1/[2(F
o2) + (0.0858P)2
+ 0.0511P]
whereP= (Fo2+ 2Fc2)/3
(/)max= 0.001
max= 0.12 e AÊÿ3
min=ÿ0.14 e AÊÿ3
Extinction correction:SHELXL97 Extinction coef®cient: 0.0052 (12) Absolute structure: Flack (1983) Flack parameter = not reliably
determined [ÿ0.7 (6)]
Table 1
Selected geometric parameters (). C5ÐC4ÐC10 89.24 (16)
C4ÐC5ÐC11 84.03 (15) C11ÐC10ÐC4C10ÐC11ÐC5 84.44 (15)89.02 (15)
All data was employed in the re®nement with the exception of the (1,1,2) re¯ection which had strongly negative intensity. During the course of the re®nement, the disorder in the C(Me)2(CH2)3backbone
of the seven-membered ring became apparent. This disorder was modelled by introducing two (CH2)3moieties anchored at both ends
to the undisordered fragment of the molecule (C71/C81/C91 and C72/ C82/C92), the occupation factors of which were allowed to re®ne. While bond-length restraints were introduced during the initial phases of the disordered re®nement to minimize the likelihood of a divergent re®nement, these proved to be unnecessary and were removed during the ®nal cycles of least-squares re®nement. Though under this model the two methyl groups of the C(Me)2moiety may
also be expected to be disordered, a model taking this aspect into account failed to yield lower residuals. H atoms were placed in geometrically idealized positions and given a common displacement parameter by class (methyl group H atoms, all others), which were allowed to re®ne. The ®nal value ofUiso(H) for the methyl group H atoms is 0.141 (4) AÊ2, and for all other H atoms is 0.095 (3) AÊ2.
Data collection: XSCANS (Siemens, 1996); cell re®nement:
XSCANS; data reduction: XSCANS; program(s) used to solve structure: SHELXS97 (Sheldrick, 1997); program(s) used to re®ne structure: SHELXL97 (Sheldrick, 1997); molecular graphics:
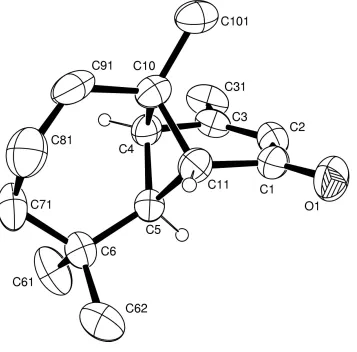
ORTEP-3 (Farrugia, 1997). Figure 1
The authors wish to acknowledge partial support of this work by the Oregon State University Marine/Freshwater Biomedical Sciences Center through grant No. P30-ES03850.
References
Baig, M. A., Banthorpe, D. V. & Gutowski, J. A. (1989).Fitoterapia,60, 373± 375.
Farrugia, L. J. (1997).J. Appl. Cryst.30, 565. Flack, H. D. (1983).Acta Cryst.A39, 876±881.
North, A. C. T., Phillips, D. C. & Mathews, F. S. (1968).Acta Cryst.A24, 351± 359.
Sheldrick, G. M. (1997). SHELXS97 and SHELXL97. University of GoÈttingen, Germany.
Siemens (1990).XEMP.Version 4.3. Siemens Analytical X-ray Instruments Inc., Madison, Wisconsin, USA.
Siemens (1996).XSCANS. Version 2.2. Siemens Analytical X-ray Instruments Inc., Madison, Wisconsin, USA.
Uchio, Y. (1978).Tetrahedron,34, 2893±2899.
Uchio, Y., Matsuo, A., Eguchi, S. Nakayama, M. & Hayashi, S. (1977).
Tetrahedron Lett.pp. 1191±1194.
White, J. D. & Lee, N. C. (1997).Advances in Organic Chemistry, Vol. 6, edited by B. Halton, pp. 1±34. Hampton Hill, England: Jai Press.
supporting information
sup-1
Acta Cryst. (2001). E57, o99–o101supporting information
Acta Cryst. (2001). E57, o99–o101 [doi:10.1107/S1600536800020079]
Vulgarone B
Alex F. T. Yokochi, James D. White and George Sturtz
S1. Comment
Continuing our structural and synthetic studies of the effects of ring strain on the properties of carbocyclic natural
products (White & Lee, 1997), we report here the structure of vulgarone B, (I). This sesquiterpene was isolated as the
major terpenoid component (ca 40%) of the essential oil of Artemisia douglasiana Bess., a plant that occurs widely in
Western Oregon. An extract of this plant has been found to possess potent insecticidal and gastropod repellent activity.
Previously, vulgarone B was isolated along with its isomer vulgarone A as a minor constituent of the oil of the medicinal
plant Chrysanthemum vulgare (L.) Bernh, [Tanacetum vulgare (L)] (Uchio et al., 1977). It has also been obtained from
the volatile oil of cultivars of Santolina chamaecyparcissus L. (Baig et al., 1989).
The structure of vulgarone B, including its absolute configuration, had earlier been deduced from spectroscopic
evidence and by chemical correlation with the related sesquiterpene (+)-α-longipinene (Uchio, 1978). The results from
our X-ray single-crystal diffraction experiment confirm the assigned structure. We also attempted to determine the
absolute configuration of vulgarone B from the diffraction data, but, due to the small magnitude of the anomalous
scattering components, the refined value of the absolute structure parameter could not be reliably determined. This is
reflected in the fact that the model shown in Fig. 1 yields a value of -0.7 (6) for this parameter, whereas the inverted
model yields a value of 1.7 (6). Further, since the absolute structure derived from the model shown is identical to the
previously deduced configuration for this molecule (Uchio, 1978), we find it reasonable to conclude that this is the true
configuration of vulgarone B.
All the observed intramolecular distances and angles are well within the expected values. The most interesting feature
of the molecule is the four-membered ring, in which all four atoms are stereogenic. Opposite corners of this ring are the
bridgehead C atoms that are components of two other rings, i.e. a seven-membered cycle and a six-membered ring. The
six-membered ring contains a C═C\sb C═O moiety, which should force five of the six atoms in the ring to be planar. This
is found to be correct; atoms O1, C1, C2, C3, C31, C4 and C11 are nearly coplanar, with a maximum deviation from the
mean plane of 0.028 (2) Å by atom C2. This geometric constraint also causes a slight distortion of the four-membered
ring, in which the bridgehead C atoms of the six-membered ring have moved toward each other. This is reflected in the
C\sb C\sb C angles within the ring, which are identical pairwise at about 84 and 89° as shown in Table 1.
S2. Experimental
Crystals of the title compound were obtained by sublimation of a bulk sample under ambient pressure and temperature.
Due to the high vapor pressure of the compound, it also sublimes during the diffraction experiment. Thus, in order to
minimize the rate of sublimation, the crystal was completely encapsulated in a thin layer of epoxy glue. This strategy was
supporting information
sup-2
Acta Cryst. (2001). E57, o99–o101S3. Refinement
All data was employed in the refinement with the exception of the (1,1,2) reflection which had strongly negative
intensity. During the course of the refinement, the disorder in the C(Me)2(CH2)3 backbone of the seven-membered ring
became apparent. This disorder was modelled by introducing two (CH2)3 moieties anchored at both ends to the
undisordered fragment of the molecule (C71/C81/C91 and C72/C82/C92), the occupation factors of which were allowed
to refine. While bond-length restraints were introduced during the initial phases of the disordered refinement to minimize
the likelyhood of a divergent refinement, these proved to be unnecessary and were removed during the final cycles of
least-squares refinement. Though under this model the two methyl groups of the C(Me)2 moiety may also be expected to
be disordered, a model taking this aspect into account failed to yield lower residuals. H atoms were placed in
geometrically idealized positions and given a common displacement parameter by class (methyl group H atoms, all
others), which were allowed to refine. The final value of Uiso(H) for the methyl group H atoms is 0.141 (4) Å2, and for all
[image:5.610.132.489.262.614.2]other H atoms is 0.095 (3) Å2.
Figure 1
View of (I) illustrating the numbering scheme (30% probability displacement ellipsoids). The minor fraction of the
supporting information
sup-3
Acta Cryst. (2001). E57, o99–o101(I)
Crystal data
C15H22O
Mr = 218.33
Orthorhombic, P212121
a = 6.628 (1) Å
b = 10.218 (1) Å
c = 19.889 (1) Å
V = 1347.0 (3) Å3
Z = 4
F(000) = 480
Dx = 1.077 Mg m−3
Cu Kα radiation, λ = 1.54178 Å Cell parameters from 99 reflections
θ = 8.0–27.3°
µ = 0.50 mm−1
T = 293 K Block, colorless 0.4 × 0.4 × 0.3 mm
Data collection
Serial
diffractometer
ω/2θ scans
Absorption correction: empirical (using intensity measurements)
via ψ scans ((North et al., 1968) using XEMP
(Siemens, 1990)
Tmin = 0.827, Tmax = 0.866
2270 measured reflections
1911 independent reflections 1666 reflections with I > 2σ(I)
Rint = 0.041
θmax = 67.6°, θmin = 4.5°
h = −6→7
k = −12→12
l = −23→23
3 standard reflections every 97 reflections intensity decay: 1%
Refinement
Refinement on F2
Least-squares matrix: full
R[F2 > 2σ(F2)] = 0.050
wR(F2) = 0.132
S = 1.07 1911 reflections 176 parameters 0 restraints
H-atom parameters constrained
w = 1/[σ2(F
o2) + (0.0858P)2 + 0.0511P]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max = 0.001
Δρmax = 0.12 e Å−3
Δρmin = −0.14 e Å−3
Extinction correction: SHELXL97, Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4
Extinction coefficient: 0.0052 (12) Absolute structure: Flack (1983)
Absolute structure parameter: not reliably determined [−0.7 (6)]
Special details
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq Occ. (<1)
O1 0.7435 (3) 0.5483 (2) 0.46305 (11) 0.1032 (7)
C1 0.5760 (3) 0.5489 (2) 0.43741 (11) 0.0706 (6)
C2 0.4529 (4) 0.6664 (2) 0.43026 (12) 0.0739 (6)
H2 0.5013 0.7476 0.4439 0.095 (3)*
C3 0.2689 (4) 0.6543 (2) 0.40359 (10) 0.0685 (6)
C31 0.1219 (5) 0.7633 (3) 0.39466 (13) 0.0932 (8)
supporting information
sup-4
Acta Cryst. (2001). E57, o99–o101H31B 0.0735 0.7637 0.3492 0.141 (4)*
H31C 0.1871 0.8452 0.4042 0.141 (4)*
C4 0.2094 (3) 0.5195 (2) 0.38143 (10) 0.0654 (6)
H4 0.0766 0.5116 0.3602 0.095 (3)*
C5 0.3913 (3) 0.4655 (2) 0.34020 (10) 0.0644 (6)
H5 0.4690 0.5392 0.3223 0.095 (3)*
C6 0.3674 (4) 0.3624 (2) 0.28532 (13) 0.0837 (7)
C61 0.2627 (7) 0.4305 (4) 0.22528 (16) 0.1334 (14)
H61A 0.2533 0.3705 0.1883 0.141 (4)*
H61B 0.3401 0.5056 0.2119 0.141 (4)*
H61C 0.1298 0.4578 0.2384 0.141 (4)*
C62 0.5757 (6) 0.3206 (4) 0.26353 (18) 0.1190 (12)
H62A 0.5649 0.2598 0.2269 0.141 (4)*
H62B 0.6437 0.2794 0.3005 0.141 (4)*
H62C 0.6510 0.3959 0.2493 0.141 (4)*
C71 0.2145 (19) 0.2555 (10) 0.2987 (6) 0.098 (3) 0.565 (14)
H71A 0.0800 0.2919 0.2945 0.095 (3)* 0.565 (14)
H71B 0.2291 0.1881 0.2647 0.095 (3)* 0.565 (14)
C81 0.2355 (19) 0.1934 (7) 0.3674 (4) 0.105 (3) 0.565 (14)
H81A 0.1565 0.1136 0.3690 0.095 (3)* 0.565 (14)
H81B 0.3757 0.1706 0.3752 0.095 (3)* 0.565 (14)
C91 0.167 (2) 0.2825 (15) 0.4200 (8) 0.094 (5) 0.565 (14)
H91A 0.1715 0.2321 0.4613 0.095 (3)* 0.565 (14)
H91B 0.0255 0.2982 0.4108 0.095 (3)* 0.565 (14)
C72 0.295 (3) 0.2293 (16) 0.3175 (8) 0.106 (4) 0.435 (14)
H72A 0.2717 0.1662 0.2819 0.095 (3)* 0.435 (14)
H72B 0.4011 0.1954 0.3461 0.095 (3)* 0.435 (14)
C82 0.1036 (19) 0.2432 (10) 0.3587 (5) 0.096 (4) 0.435 (14)
H82A 0.0323 0.1603 0.3586 0.095 (3)* 0.435 (14)
H82B 0.0170 0.3078 0.3376 0.095 (3)* 0.435 (14)
C92 0.146 (3) 0.2866 (13) 0.4358 (11) 0.075 (4) 0.435 (14)
H92A 0.0186 0.2941 0.4596 0.095 (3)* 0.435 (14)
H92B 0.2262 0.2202 0.4580 0.095 (3)* 0.435 (14)
C10 0.2574 (3) 0.4183 (2) 0.43794 (11) 0.0682 (6)
C101 0.2191 (4) 0.4569 (3) 0.51137 (11) 0.0833 (7)
H10A 0.0786 0.4764 0.5174 0.141 (4)*
H10B 0.2981 0.5327 0.5223 0.141 (4)*
H10C 0.2567 0.3857 0.5403 0.141 (4)*
C11 0.4774 (3) 0.4276 (2) 0.41107 (11) 0.0665 (6)
H11 0.5576 0.3472 0.4132 0.095 (3)*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
O1 0.0654 (11) 0.1316 (16) 0.1126 (14) −0.0036 (11) −0.0180 (10) −0.0206 (12)
C1 0.0560 (12) 0.0910 (15) 0.0649 (11) −0.0036 (11) −0.0026 (10) −0.0031 (11)
C2 0.0859 (17) 0.0690 (13) 0.0668 (12) −0.0056 (12) −0.0046 (13) −0.0065 (10)
supporting information
sup-5
Acta Cryst. (2001). E57, o99–o101C31 0.115 (2) 0.0872 (16) 0.0778 (14) 0.0329 (16) 0.0104 (15) 0.0097 (13)
C4 0.0529 (11) 0.0794 (13) 0.0638 (11) −0.0003 (10) −0.0059 (10) 0.0037 (10)
C5 0.0616 (12) 0.0661 (11) 0.0655 (11) −0.0043 (11) −0.0018 (10) −0.0037 (9)
C6 0.0855 (16) 0.0822 (15) 0.0833 (15) −0.0123 (14) 0.0031 (14) −0.0230 (12)
C61 0.165 (4) 0.144 (3) 0.0909 (19) −0.007 (3) −0.036 (2) −0.037 (2)
C62 0.120 (3) 0.127 (2) 0.109 (2) 0.008 (2) 0.021 (2) −0.046 (2)
C71 0.116 (8) 0.077 (4) 0.101 (6) −0.025 (4) −0.025 (5) −0.013 (4)
C81 0.120 (6) 0.070 (3) 0.125 (6) −0.017 (4) −0.007 (5) 0.013 (3)
C91 0.067 (5) 0.125 (8) 0.091 (10) −0.014 (4) 0.015 (5) 0.028 (5)
C72 0.110 (11) 0.103 (9) 0.105 (9) −0.024 (7) 0.000 (6) −0.036 (7)
C82 0.089 (6) 0.075 (4) 0.123 (7) −0.020 (4) 0.002 (5) −0.014 (4)
C92 0.099 (9) 0.059 (5) 0.068 (7) −0.015 (4) 0.025 (4) 0.010 (3)
C10 0.0576 (12) 0.0760 (13) 0.0709 (12) 0.0017 (11) 0.0026 (11) 0.0119 (10)
C101 0.0750 (14) 0.1046 (17) 0.0703 (13) 0.0082 (15) 0.0099 (11) 0.0212 (12)
C11 0.0563 (12) 0.0705 (12) 0.0727 (12) 0.0053 (10) −0.0030 (10) 0.0022 (9)
Geometric parameters (Å, º)
O1—C1 1.222 (3) C6—C71 1.514 (12)
C1—C2 1.458 (3) C6—C61 1.546 (5)
C1—C11 1.496 (3) C6—C72 1.579 (17)
C2—C3 1.336 (4) C71—C81 1.512 (16)
C3—C31 1.491 (3) C81—C91 1.459 (17)
C3—C4 1.498 (3) C91—C10 1.553 (15)
C4—C5 1.559 (3) C72—C82 1.52 (2)
C4—C10 1.560 (3) C82—C92 1.62 (2)
C5—C6 1.525 (3) C92—C10 1.537 (15)
C5—C11 1.569 (3) C10—C101 1.534 (3)
C6—C62 1.509 (5) C10—C11 1.556 (3)
O1—C1—C2 123.6 (2) C5—C6—C72 109.6 (5)
O1—C1—C11 122.6 (2) C61—C6—C72 124.4 (7)
C2—C1—C11 113.78 (19) C81—C71—C6 113.6 (8)
C3—C2—C1 118.2 (2) C91—C81—C71 111.0 (10)
C2—C3—C31 125.1 (2) C81—C91—C10 127.1 (11)
C2—C3—C4 116.2 (2) C82—C72—C6 113.2 (13)
C31—C3—C4 118.7 (2) C72—C82—C92 113.1 (12)
C3—C4—C5 106.07 (18) C10—C92—C82 110.5 (12)
C3—C4—C10 110.09 (16) C101—C10—C92 99.9 (8)
C5—C4—C10 89.24 (16) C101—C10—C91 112.6 (6)
C6—C5—C4 122.73 (19) C92—C10—C91 12.9 (13)
C6—C5—C11 120.70 (19) C101—C10—C11 117.80 (19)
C4—C5—C11 84.03 (15) C92—C10—C11 119.7 (8)
C62—C6—C71 117.3 (5) C91—C10—C11 109.7 (6)
C62—C6—C5 107.8 (2) C101—C10—C4 118.83 (19)
C71—C6—C5 116.2 (5) C92—C10—C4 117.5 (8)
C62—C6—C61 108.4 (3) C91—C10—C4 110.4 (6)
supporting information
sup-6
Acta Cryst. (2001). E57, o99–o101C5—C6—C61 106.8 (2) C1—C11—C10 109.86 (19)
C62—C6—C72 98.7 (7) C1—C11—C5 105.62 (18)