organic papers
Acta Cryst.(2007). E63, o1475–o1477 doi:10.1107/S1600536807008896 Engel and Ferrence C
4H10N2O
o1475
Acta Crystallographica Section EStructure Reports Online
ISSN 1600-5368
2-Methylpropionohydrazide
Sharon E. Engel and Gregory M. Ferrence*
CB 4160, Department of Chemistry, Illinois State University, Normal, IL 61790, USA
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study T= 100 K
Mean(C–C) = 0.003 A˚ Rfactor = 0.073 wRfactor = 0.191
Data-to-parameter ratio = 16.9
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
Received 20 February 2007 Accepted 22 February 2007
#2007 International Union of Crystallography All rights reserved
In the title structure, C4H10N2O, the molecules stack with
intermolecular N—H O hydrogen bonds along the b axis and additional intermolecular N—H N hydrogen bonds form a network orthogonal to thecaxis.
Comment
As part of our investigation into the synthesis of dialkyl- and aryl-substituted triazoles (Jerniganet al., 2007), compound (I) was isolated as a minor by-product in the synthesis of 3,5-diisopropyl-1,2,4-triazole. The title compound, (I), has been known for over 40 years (Rabini & Vita 1965); however, this is the first report of its X-ray crystal structure.
A Mogul (Bruno et al. 2004) geometry check showed all non-H bond angles and distances to be normal. The two most similar molecules identified in the Cambridge Structural Database (Version 5.28; Allen, 2002) are cyclopropane-carboxylic acid hydrazide, (II) (Chesnut & Marsh, 1958), and
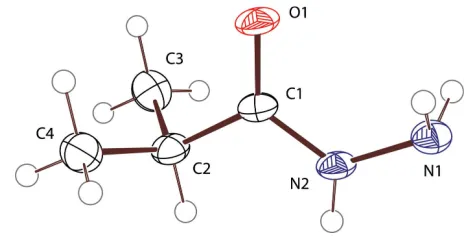
n-nonanoic acid hydrazide (Jensen & Lingafelter, 1961). Comparison of these two molecules with the title compound show corresponding bond distances to be nearly indis-tinguishable and corresponding angles to differ by less than two degrees. The obvious exception are the bond angles about C2, where the cyclopropyl-ring-imposed 58.8 C3—C2—C4
bond angle of (II) is substantially less than the 111.5 (2)angle
[image:1.610.213.448.605.724.2]observed in (I). Likewise, the C1—C2—C3 and C1—C2—C4 angles are larger in (II) than in (I).
Figure 1
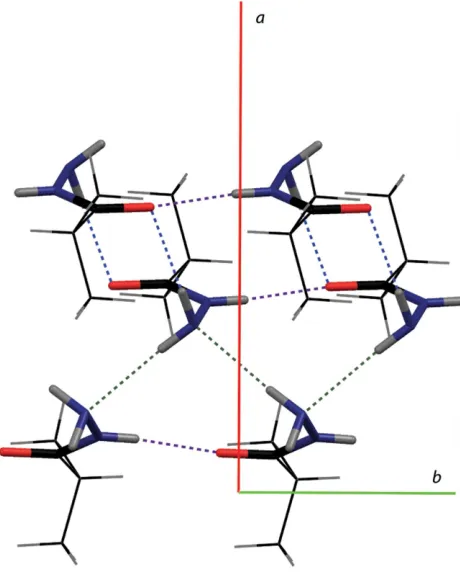
Compound (I) crystallizes with an extensive network of intermolecular hydrogen bonding (Table 1). The molecules stack with N2—H2N O1ihydrogen bonds along thebaxis (Fig. 2; purple dashes), with moderate 2.831 (2) A˚ D A
separations. N1—H1A N1iii hydrogen bonds across the a -glide planes (Fig. 2; green dashes) pair these stacks together with 3.158 (3) A˚ D Aseparations. These pairs of stacks are further joined to neighboring pairs of stacks through N1— H1B O1iihydrogen bonds (Fig. 2; blue dashes), the pairs of molecules being related by an inversion center. Overall, a two-dimensional network of intermolecularly hydrogen-bonded molecules is formed orthogonal to the c axis (Fig. 3). The hydrophobic isopropyl groups are oriented away from the hydrogen-bonding network, forming layers along the c axis connected only by van der Waals contacts (Fig. 4). This hydrogen-bonding network is essentially the same as that observed in (II) (N1 N10= 3.16, N1 O0= 3.26 and N2 O1 = 2.94 A˚ ). It is worth noting that the 3.078 (3) A˚ N1 O0
D A separations observed for (I) are shorter than those observed for (II) by 0.18 A˚ . This suggests that the modest geometric alterations between isopropyl and cyclopropyl significantly impacted these hydrogen bonds. This is consistent with the conclusion that the N1 O0bond must be considered
a very weak link (Chesnut & Marsh, 1958).
Experimental
The synthesis of 3,5-diisopropyltriazole was carried out in a manner analogous to the literature procedure for the synthesis of 3-tert
-butyl-5-methyltriazole (Jerniganet al., 2007; Pe´rezet al., 1983). During the purification of 3,5-diisopropyltriazole by sublimation under vacuum at 383 K, two separate rings of (I) were deposited on the wall of the sublimation cold-finger. The top band consisted of colorless clear crystals from which one was selected for the single-crystal X-ray diffraction experiment. Once the structure was determined to be that of the title compound, a compound known for over 40 years (Rabini & Vita 1965), no further characterization was carried out.
organic papers
o1476
Engel and Ferrence C [image:2.610.345.523.69.361.2]4H10N2O Acta Cryst.(2007). E63, o1475–o1477
Figure 2
[image:2.610.55.285.72.361.2]A view of the principal hydrogen bonds (dashed lines) in (I).
Figure 3
A view orthogonal to the ab plane, showing the two-dimensional arrangement of hydrogen bonds (dashed lines).
Figure 4
[image:2.610.316.564.414.615.2]Crystal data
C4H10N2O
Mr= 102.08
Orthorhombic,Pbca a= 11.0189 (19) A˚
b= 4.8781 (9) A˚
c= 21.677 (4) A˚
V= 1165.2 (4) A˚3
Z= 8
MoKradiation
= 0.09 mm1
T= 100 (2) K 0.590.460.18 mm
Data collection
Bruker SMART APEX CCD diffractometer
Absorption correction: none 8412 measured reflections
1755 independent reflections 1246 reflections withI> 2(I)
Rint= 0.063
Refinement
R[F2> 2(F2)] = 0.073
wR(F2) = 0.191
S= 1.12 1755 reflections
104 parameters
All H-atom parameters refined
max= 0.44 e A˚
3
min=0.29 e A˚
3
Table 1
Hydrogen-bond geometry (A˚ ,).
D—H A D—H H A D A D—H A
N2—H2N O1i
0.85 (3) 1.98 (3) 2.831 (2) 175 (3) N1—H1B O1ii 0.92 (3) 2.17 (3) 3.078 (3) 169 (2) N1—H1A N1iii
0.90 (3) 2.26 (3) 3.158 (3) 172 (2)
Symmetry codes: (i)x;yþ1;z; (ii)xþ1;yþ1;zþ1; (iii)xþ1 2;y
1 2;z.
All H atoms were identified in difference Fourier maps and refined with isotropic displacement parameters.
Data collection:SMART(Bruker, 2002); cell refinement: SAINT-Plus(Bruker, 2003); data reduction:SAINT-Plus; program(s) used to solve structure:SIR2004(Burlaet al., 2005); program(s) used to refine structure: SHELXL97 (Sheldrick, 1997); molecular graphics:
ORTEP-3 for Windows(Farrugia, 1997) andMERCURY(Macraeet
al., 2006); software used to prepare material for publication:WinGX
(Farrugia, 1997) andpublCIF(Westrip, 2007).
GMF gratefully acknowledges the Research Corporation (Grant CC6205) and the National Science Foundation (NSF; Grant CHE-0348158) for support. SEE thanks the NSF for a GK-12 Graduate Fellowship. GMF thanks Matthias Zeller of Youngstown State University Structure & Chemical Instru-mentation Facility for the data collection and useful discus-sions. The diffractometer was funded by NSF grant 0087210, Ohio Board of Regents grant CAP-491, and YSU.
References
Allen, F. H. (2002).Acta Cryst.B58, 380–388.
Bruker (2002). SMART for WNT/2000. Version 5.630. Bruker AXS Inc, Madison, Wisconsin, USA.
Bruker (2003). SAINT-Plus. Version 6.45. Bruker AXS Inc, Madison, Wisconsin, USA.
Bruno, I. J., Cole, J. C., Kessler, M., Luo, J., Motherwell, W. D. S., Purkis, L. H., Smith, B. R., Taylor, R., Cooper, R. I., Harris, S. E. & Orpen, A. G. (2004).J. Chem. Inf. Comput. Sci.44, 2133–2144.
Burla, M. C., Caliandro, R., Camalli, M., Carrozzini, B., Cascarano, G. L., De Caro, L., Giacovazzo, C., Polidori, G. & Spagna, R. (2005).J. Appl. Cryst.38, 381–388.
Chesnut, D. B. & Marsh, R. E. (1958).Acta Cryst.11, 413–419. Farrugia, L. J. (1997).J. Appl. Cryst.30, 565.
Jensen, L. H. & Lingafelter, E. C. (1961).Acta Cryst.14, 507–520.
Jernigan, F. E. III, Sieracki, N. A., Taylor, M. T., Jenkins, A. S., Engel, S. E., Rowe, B. W., Jove´, F. A., Yap, G. P. A., Papish, E. T. & Ferrence, G. F. (2007).
Inorg. Chem.46, 360–362.
Macrae, C. F., Edgington, P. R., McCabe, P., Pidcock, E., Shields, G. P., Taylor, R., Towler, M. & van de Streek, J. (2006).J. Appl. Cryst.39, 453–457. Pe´rez, M. A., Dorado, C. A. & Soto, J. L. (1983).Synthesis,6, 483–486. Rabini, T. & Vita, G. (1965).J. Org. Chem.30, 2486–2488.
Sheldrick, G. M. (1997).SHELXL97. University of Go¨ttingen, Germany. Westrip, S. P. (2007).publCIF. In preparation.
organic papers
Acta Cryst.(2007). E63, o1475–o1477 Engel and Ferrence C
supporting information
sup-1 Acta Cryst. (2007). E63, o1475–o1477
supporting information
Acta Cryst. (2007). E63, o1475–o1477 [https://doi.org/10.1107/S1600536807008896]
2-Methylpropionohydrazide
Sharon E. Engel and Gregory M. Ferrence
2-Methylpropionohydrazide
Crystal data
C4H10N2O
Mr = 102.08
Orthorhombic, Pbca
Hall symbol: -P 2ac 2ab
a = 11.0189 (19) Å
b = 4.8781 (9) Å
c = 21.677 (4) Å
V = 1165.2 (4) Å3
Z = 8
F(000) = 448
Dx = 1.165 Mg m−3
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 9311 reflections
θ = 5.3–61.0°
µ = 0.09 mm−1
T = 100 K Plate, colourless 0.59 × 0.46 × 0.18 mm
Data collection
Bruker SMART APEX CCD diffractometer
Radiation source: sealed tube Graphite monochromator
ω scans
8412 measured reflections 1755 independent reflections
1246 reflections with I > 2σ(I)
Rint = 0.063
θmax = 30.5°, θmin = 1.9°
h = −15→14
k = −6→6
l = −30→30
Refinement
Refinement on F2 Least-squares matrix: full
R[F2 > 2σ(F2)] = 0.073
wR(F2) = 0.191
S = 1.12 1755 reflections 104 parameters
0 restraints
All H-atom parameters refined
w = 1/[σ2(F
o2) + (0.0748P)2 + 0.9877P] where P = (Fo2 + 2Fc2)/3
(Δ/σ)max < 0.001 Δρmax = 0.44 e Å−3 Δρmin = −0.29 e Å−3
Special details
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
N1 0.34101 (18) 0.7903 (4) 0.48473 (8) 0.0254 (4)
supporting information
sup-2 Acta Cryst. (2007). E63, o1475–o1477
C1 0.42395 (19) 0.6629 (4) 0.58377 (10) 0.0238 (4)
O1 0.41985 (15) 0.4143 (3) 0.57242 (7) 0.0290 (4)
C2 0.4730 (2) 0.7685 (4) 0.64454 (10) 0.0266 (5)
C3 0.6052 (2) 0.6864 (6) 0.65061 (12) 0.0348 (6)
C4 0.3968 (2) 0.6531 (5) 0.69710 (11) 0.0339 (5)
H1A 0.292 (3) 0.643 (6) 0.4885 (12) 0.033 (7)*
H1B 0.408 (3) 0.734 (6) 0.4627 (13) 0.037 (7)*
H2N 0.392 (2) 1.021 (7) 0.5539 (12) 0.032 (7)*
H2A 0.469 (2) 0.966 (6) 0.6446 (10) 0.028 (6)*
H3A 0.611 (3) 0.482 (7) 0.6498 (13) 0.037 (7)*
H3B 0.640 (3) 0.748 (6) 0.6876 (13) 0.038 (7)*
H3C 0.649 (3) 0.755 (7) 0.6196 (15) 0.048 (8)*
H4A 0.403 (2) 0.447 (6) 0.6966 (12) 0.038 (7)*
H4B 0.426 (2) 0.708 (5) 0.7355 (12) 0.030 (6)*
H4C 0.309 (3) 0.696 (6) 0.6935 (13) 0.040 (7)*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
N1 0.0269 (9) 0.0188 (8) 0.0306 (9) −0.0019 (7) −0.0010 (7) −0.0016 (7)
N2 0.0290 (9) 0.0126 (8) 0.0315 (9) 0.0000 (7) −0.0022 (7) −0.0021 (6)
C1 0.0244 (10) 0.0152 (9) 0.0317 (10) 0.0002 (7) 0.0015 (8) −0.0005 (7)
O1 0.0377 (9) 0.0113 (7) 0.0380 (8) 0.0000 (6) −0.0036 (6) −0.0019 (6)
C2 0.0317 (11) 0.0166 (9) 0.0314 (10) −0.0011 (8) −0.0020 (8) −0.0005 (7)
C3 0.0327 (13) 0.0374 (14) 0.0343 (12) −0.0053 (10) −0.0013 (10) −0.0035 (10)
C4 0.0362 (13) 0.0321 (12) 0.0333 (11) 0.0007 (10) 0.0036 (9) −0.0024 (9)
Geometric parameters (Å, º)
N1—N2 1.415 (2) N2—H2N 0.85 (3)
N2—C1 1.328 (3) C2—H2A 0.96 (3)
C1—O1 1.238 (2) C3—H3A 1.00 (3)
C1—C2 1.514 (3) C3—H3B 0.94 (3)
C2—C3 1.516 (3) C3—H3C 0.89 (3)
C2—C4 1.523 (3) C4—H4A 1.01 (3)
N1—H1A 0.90 (3) C4—H4B 0.93 (3)
N1—H1B 0.92 (3) C4—H4C 1.00 (3)
N1—N2—C1 123.28 (17) H3A—C3—H3C 109 (3)
N2—C1—O1 122.9 (2) H3B—C3—H3C 108 (3)
O1—C1—C2 121.26 (19) C2—C4—H4A 108.7 (16)
N2—C1—C2 115.86 (18) C2—C4—H4B 112.0 (16)
C1—C2—C3 109.18 (18) H4A—C4—H4B 106 (2)
C1—C2—C4 109.16 (18) C2—C4—H4C 113.7 (16)
C3—C2—C4 111.5 (2) H4A—C4—H4C 106 (2)
C1—C2—H2A 109.0 (14) H4B—C4—H4C 110 (2)
C3—C2—H2A 107.9 (16) N2—N1—H1A 107.7 (17)
supporting information
sup-3 Acta Cryst. (2007). E63, o1475–o1477
C2—C3—H3A 108.9 (17) H1A—N1—H1B 107 (2)
C2—C3—H3B 112.4 (18) C1—N2—H2N 119.3 (19)
H3A—C3—H3B 108 (2) N1—N2—H2N 117.4 (19)
C2—C3—H3C 111 (2)
O1—C1—C2—C3 62.2 (3) N2—C1—C2—C4 120.4 (2)
N2—C1—C2—C3 −117.4 (2) O1—C1—N2—N1 −0.9 (3)
O1—C1—C2—C4 −60.0 (3) C2—C1—N2—N1 178.73 (18)
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
N2—H2N···O1i 0.85 (3) 1.98 (3) 2.831 (2) 175 (3)
N1—H1B···O1ii 0.92 (3) 2.17 (3) 3.078 (3) 169 (2)
N1—H1A···N1iii 0.90 (3) 2.26 (3) 3.158 (3) 172 (2)
![Methyl ({[(4E) 1,3 dimethyl 2,6 diphenylpiperidin 4 ylidene]amino}oxy)acetate](data:image/gif;base64,R0lGODlhAQABAIAAAP///wAAACH5BAEAAAAALAAAAAABAAEAAAICRAEAOw==)