Causing CHARGE Syndrome
abstract
+We report the case of a 15-year-old girl who presented to a pediatric endocrinology clinic for delayed puberty with no signs of secondary sexual development. Her past medical history was significant for bilateral colobomas, inner-ear anomalies, hearing loss, and anos-mia. Genetic testing revealed a novel de novo mutation in theCHD7
gene, one of the causative genes in CHARGE syndrome (coloboma, heart disease, choanal atresia, retarded growth and development and/or central nervous system anomalies, genital anomalies and/or hypogonadism, and ear anomalies and/or deafness). We review the distinction between hypogonadotrophic hypogonadism and hypergonadotrophic hypogonadism and discuss the availability of molecular genetic testing for idiopathic hypogonadotrophic hy-pogonadism. CHD7 mutations have also been found in some pa-tients with Kallmann syndrome, hypogonadotrophic hypogonadism, and anosmia, and we discuss the overlap between this syndrome and CHARGE syndrome. With the increased availability of genetic testing for a variety of disorders, it is important for pediatricians to become familiar with interpreting genetic test results. Finally, we illustrate that Bayes’ theorem is a useful statistical tool for inter-preting novel missense mutations of unknown significance.
Pediatrics2010;126:e1594–e1598
AUTHORS:Andrew Dauber, MD,a,bJoel N. Hirschhorn, MD,
PhD,a,c,d,eJonathan Picker, MBChB, PhD,c,fThomas A.
Maher, MS,gand Aubrey Milunsky, MBBCh, DScg
Divisions ofaEndocrinology andcGenetics andfDepartment of
Child Psychiatry, Children’s Hospital Boston, Boston, Massachusetts;bClinical Investigator Training Program,
Harvard/MIT Health Sciences and Technology-Beth Israel Deaconess Medical Center, Boston, Massachusetts;dDepartment
of Genetics, Harvard Medical School, Boston, Massachusetts;
eBroad Institute, Boston, Massachusetts; andgCenter for Human
Genetics, Boston University School of Medicine, Boston, Massachusetts
KEY WORDS
CHD7, CHARGE, Kallmann syndrome, hypogonadotrophic hypogonadism, delayed puberty
ABBREVIATIONS
IHH—idiopathic hypogonadotrophic hypogonadism CHARGE—coloboma, heart disease, choanal atresia, retarded growth and development and/or central nervous system anomalies, genital anomalies and/or hypogonadism, and ear anomalies and/or deafness
KS—Kallmann syndrome
nIHH—normosmic idiopathic hypogonadotrophic hypogonadism www.pediatrics.org/cgi/doi/10.1542/peds.2010-0164
doi:10.1542/peds.2010-0164 Accepted for publication Jul 29, 2010
Address correspondence to Andrew Dauber, MD, Division of Endocrinology, CLS 16, Children’s Hospital Boston, 300 Longwood Ave, Boston, MA 02115. E-mail: andrew.dauber@childrens. harvard.edu
PEDIATRICS (ISSN Numbers: Print, 0031-4005; Online, 1098-4275). Copyright © 2010 by the American Academy of Pediatrics
Delayed puberty is a common complaint heard in pediatric endocrinology prac-tice. Recent advances in genetics re-search have described multiple genetic causes of idiopathic hypogonadotrophic hypogonadism (IHH). Pediatricians are now confronted with needing to inter-pret genetic test results including novel changes that may or may not be mutations. We report here the case of a 15-year-old girl who presented with delayed puberty and was found to have a novel mutation in theCHD7gene with phenotypic overlap between CHARGE (coloboma, heart disease, choanal atresia, retarded growth and develop-ment and/or central nervous system anomalies, genital anomalies and/or hypogonadism, and ear anomalies and/or deafness) syndrome and Kall-mann syndrome (KS).
CASE REPORT
The patient presented at the age of 15 years 6 months to our pediatric endo-crinology clinic for evaluation of de-layed puberty. She had not noted any evidence of pubertal development in-cluding breast or pubic hair develop-ment, and she had not undergone men-arche. On review of her growth chart, her birth weight was 7 lb (⬃50th per-centile), and her length was 19 inches (25th–50th percentile). Her height had been below the 5th percentile since the age of 2 years. She appeared to be
growing parallel to the growth curve until the age of 12 when her growth decelerated, which resulted in a wors-ening of her short stature. There was no evidence of a pubertal growth spurt. A review of her systems was no-table for a lifetime inability to smell.
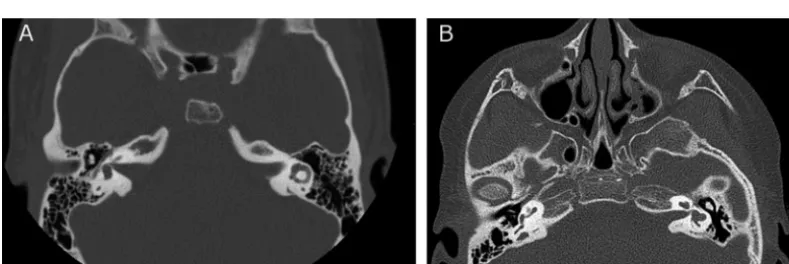
Her past medical history was signifi-cant for the presence of bilateral choroidal colobomas with a left iris coloboma that resulted in significant visual impairment. She was first noted to have hearing loss at 6 years of age and consequently wears a hearing aid. At 15 years of age, she was seen in the otolaryngology clinic for further evalu-ation of her hearing loss. Audiologic testing revealed bilateral mixed hear-ing loss. A computed tomography scan was performed (Fig 1) and showed bi-lateral inner-ear abnormalities includ-ing dysmorphic modioli, incompletely formed middle and apical turns of both cochlea, and truncated bilateral supe-rior semicircular canals. The lateral semicircular canals were patulous and truncated. The right posterior semicircular canal was small, and the right vestibular aqueduct was moder-ately enlarged. Choanal atresia was not present. The patient was otherwise healthy and cognitively normal. Her early motor milestones were mildly de-layed; she sat at 1 year and walked at 17 months. There was no family
his-tory of delayed puberty or congenital anomalies.
On physical examination, the patient’s height was 142.5 cm (⫺3.1 SDs), weight was 43 kg (⫺1.4 SDs), and BMI was 21.2 (0.3 SDs). Her examination was notable for the left iris coloboma and the presence of a hearing aid. She was at Tanner stage 1 for breast and pubic hair development.
Initial laboratory testing showed a lu-teinizing hormone value of less than the assay limit, a follicle-stimulating hormone level of 0.37 IU/L, and an es-tradiol level of 7.5 pg/mL. These results were consistent with a diagnosis of hy-pogonadotropic hypogonadism as the cause of her delayed puberty. Her insulin-like growth factor 1 level was 264 ng/mL (normal range at Tanner stage 1: 49 –342 ng/mL), and her insulin-like growth factor– binding protein 3 level was 5.4g/mL (normal range at Tanner stage 1: 1.2– 6.4 g/ mL), which is an argument against the presence of growth hormone defi-ciency. She had previously had normal thyroid function test results. A bone-age radiograph showed a delayed bone age of 12 years 6 months. Given the constellation of colobomas, inner-ear anomalies, anosmia, and hypogo-nadotrophic hypogonadism, we felt that her presentation was consistent with CHARGE syndrome. Mutational
FIGURE 1
Computed tomography scan of semicircular canals. A, Normal left semicircular canal. B, Patient’s computed tomography scan, which shows dysplastic semicircular canals (see text for detailed description).
novel mutation, c.3089A¡G, which led to a substitution of serine for aspara-gine in exon 12, p.Asn1030Ser (see
Supplemental Methods and
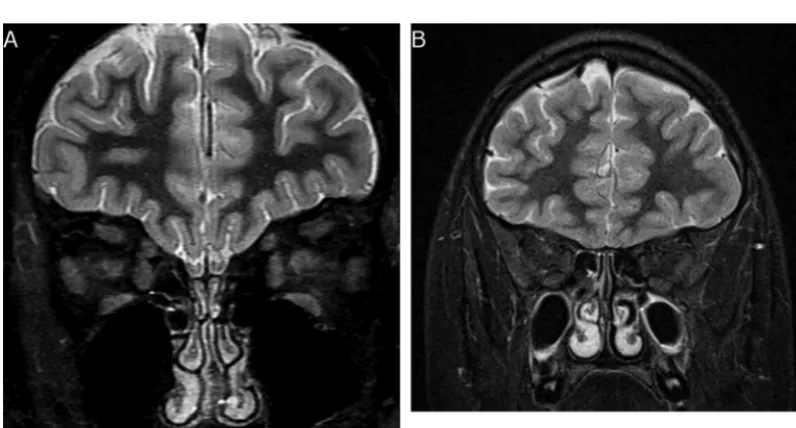
Supple-mental Fig 3). Genetic testing of her parents was performed, and neither of them was found to carry the mutation. The patient subsequently had a normal cardiac evaluation and a brain MRI (Fig 2), which showed absence of the olfac-tory bulbs and a normal pituitary gland.
DISCUSSION
Delayed puberty is defined as the absence of any secondary sexual char-acteristics at age 13 in girls and 14 in boys.1 Causes of delayed puberty
are categorized on the basis of go-nadotropin (luteinizing hormone and follicle-stimulating hormone) levels as hypergonadotrophic hypogonadism or hypogonadotrophic hypogonadism. Hypergonadotrophic hypogonadism occurs with disorders of the gonads themselves, which lead to low circulat-ing sex steroid levels and consequently elevated gonadotropin levels from lack of negative feedback. This category
in-syndrome as well as other causes of primary testicular or ovarian failure such as chemotherapy or gonadal au-toimmunity. In hypogonadotrophic hy-pogonadism, gonadotropin levels are low, which leads to a lack of stimula-tion of the gonads to produce sex ste-roids. Hypogonadotrophic hypogonad-ism can be caused by acquired disorders such as brain tumors or in-filtrative diseases of the pituitary or can be caused by genetic defects that lead to isolated gonadotropin defi-ciency or multiple pituitary hormone defects. Hypogonadotrophic hypogo-nadism can also result from chronic illness, malnutrition, or intense exer-cise. Recent advances have helped de-lineate the underlying pathophysiology of IHH. The clinical syndrome of IHH with anosmia is referred to as KS, whereas normosmic forms of IHH (nIHH) occur as well. Mutations in nu-merous genes have been found to cause⬃30% of cases of IHH.2,3
CHARGE syndrome is a clinical syn-drome named with a mnemonic that stands for coloboma, heart defects,
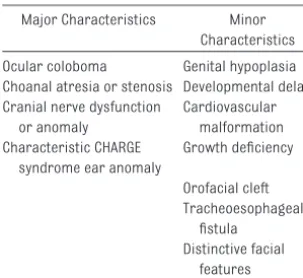
and ear anomalies. The diagnosis of definite CHARGE syndrome is based on meeting 4 major characteristics or the combination of 3 major and 3 minor characteristics (Table 1).4
Subjects who do not meet the criteria for definite CHARGE syndrome are re-ferred to as possibly or probably hav-ing CHARGE syndrome on the basis of the number of criteria met. Some au-thors have proposed revised criteria that highlight the specificity of hypo-plastic semicircular canals in the diag-nosis of CHARGE.5 In its original
de-scription, subjects with CHARGE were noted to have genital hypoplasia with micropenis in boys and labial hypopla-sia in girls. These genital anomalies were later discovered to result from low gonadotropin levels, and addi-tional subjects were described with delayed puberty that resulted from hy-pogonadotrophic hypogonadism.6,7 In
addition, one of the study reports de-scribed abnormal olfactory bulb devel-opment that led to anosmia.6TheCHD7
gene, a member of the chromodomain family, was found to be the causative
FIGURE 2
gene in CHARGE syndrome8in the
ma-jority of cases. In a large cohort,⬃60% of the subjects with the clinical diagno-sis of CHARGE syndrome were found to harbor mutations in CHD7.9 Because
of the presentation of patients with CHARGE with hypogonadotrophic hypo-gonadism along with anosmia, CHD7
was thought to be an excellent candi-date gene as a cause of KS. Subse-quently, 2 groups have reported on co-horts of subjects with KS who were found to have mutations inCHD7. Jong-mans et al10analyzed CHD7in 56
tients with KS or nIHH. Three of the pa-tients who were originally diagnosed with KS were found to have mutations inCHD7. In retrospect, these patients all had clinical features consistent with CHARGE syndrome. Kim et al11
screened a cohort of 197 subjects with KS/nIHH without the CHARGE pheno-type and 7 subjects (4 with nIHH and 3 with KS) were found to haveCHD7 mu-tations, which demonstrates the prin-ciple that mutations in a single gene can cause a spectrum of related disor-ders. CHARGE syndrome can be a diffi-cult clinical diagnosis and should be considered for all patients who present with IHH, because the clinical manifestations may be subtle.
Our patient met clinical criteria for probable CHARGE syndrome because of the presence of coloboma, inner-ear anomalies, growth retardation, hypogonadotrophic hypogonadism,
and anosmia. On further genetic test-ing, she was found to have a novel de novo heterozygous missense muta-tion inCHD7. CHARGE syndrome is in-herited in an autosomal dominant fashion, but the vast majority of cases represent de novo mutations.9
In recent years, our ability to make molecular genetic diagnoses has rapidly increased for a multitude of conditions. General pediatricians and pediatric subspecialists must be prepared to interpret the results of these tests. Novel missense muta-tions are especially difficult to inter-pret because, by definition, no one else has ever reported the effects of the mutation and the genetic alter-ation leads to a change in a single amino acid. One must decide if that change is clinically significant or if it simply reflects chance variation. In a research setting, one can attempt to design functional studies to validate the clinical significance of the muta-tion; this is clearly not practical in the clinical setting, and it is not al-ways possible in the research set-ting. In addition, there are computer programs such as PolyPhen (http:// genetics.bwh.harvard.edu/pph) or SIFT (http://sift.jcvi.org) that will pre-dict whether an individual mutation is expected to be tolerated or delete-rious. However, these programs have their limitations, as evidenced by our case, in which PolyPhen re-ported that this mutation is possibly damaging and SIFT predicted that it will be tolerated.
We propose using Bayes’ theorem to estimate the likelihood that a de novo mutation is causal for the clinical phenotype. Before applying Bayes’ theorem, one must confirm that the mutation is not present in either parent to ensure that it is truly de novo. We use our patient as an illustrative example in Table 2. In this example, once we have obtained the
genetic test results, our posttest prob-ability of our patient having CHARGE syndrome has increased to ⬎99.9% from a pretest probability of 50%. If we had been dealing with a comparable gene that only had a 10% de novo mu-tation rate and our pretest probability had only been 10%, our posttest prob-ability would have increased to 98% with finding a missense mutation. Thus, Bayes’ theorem demonstrates that for any clinical syndrome for which there is an appreciable rate of de novo mutations causing that syn-drome, finding a novel de novo mis-sense mutation should clinch the diagnosis, as long as the pretest prob-ability is not extremely low.
CONCLUSIONS
We report the case of a 15-year-old girl who presented with delayed pu-berty along with coloboma, anosmia,
TABLE 1 CHARGE Syndrome Diagnostic Criteria
Major Characteristics Minor Characteristics Ocular coloboma Genital hypoplasia Choanal atresia or stenosis Developmental delay Cranial nerve dysfunction
or anomaly
Cardiovascular malformation Characteristic CHARGE
syndrome ear anomaly
Growth deficiency Orofacial cleft Tracheoesophageal fistula Distinctive facial features
TABLE 2 Application of Bayes’ Theorem to de Novo Missense Mutations Terms
x⫽probability of discovering a de novo missense mutation inCHD7in a patient with CHARGE syndrome
y⫽pretest probability of our subject having CHARGE syndrome
z⫽probability of de novo missense mutation inCHD7in someone without CHARGE syndrome
Assumptions
60% of patients with CHARGE syndrome have a mutation inCHD7, and 90% of those are de novo; thus, termxequals 0.6⫻0.9⫽0.54 We conservatively estimate that because our patient meets diagnosis of probable CHARGE syndrome, she has a 50% chance of truly having CHARGE syndrome; thus, term
y⫽50%
The background rate of mutations in the genome is⬃1⫻10⫺8per nucleotide, and
two-thirds of all new mutations in coding regions are missense mutations; therefore, termzequals (2 chromosomes⫻8994 nucleotides in the coding region of
CHD7⫻2/3⫻1⫻10⫺8)⫽0.00012
Application of Bayes’ theorem Posttest probability of having CHARGE
syndrome⫽xy/(xy⫹z[1⫺y]) For our patient this formula equals:
(0.54⫻0.5)/([0.54⫻0.5]⫹[0.00012⫻ 0.5])⫽ ⬎99.9%
which caused CHARGE syndrome. Pe-diatricians should be aware of the increased availability of molecular
with delayed puberty. We propose us-ing Bayes’ theorem as a means of in-terpreting novel genetic results.
research project.
We thank Dr Diana Rodriguez for help with the preparation of Figs 1 and 2.
REFERENCES
1. Styne DM, Grumbach MM. Puberty: ontog-eny, neuroendocrinology, physiology, and disorders. In: Kronenberg HM, Melmed S, Polonsky KS, Larsen PR, eds.Williams Text-book of Endocrinology. Philadelphia, PA: Saunders Elsevier; 2008:969 –1166 2. Semple RK, Kemal Topaloglu A. The
re-cent genetics of hypogonadotrophic hypogonadism: novel insights and new questions. Clin Endocrinol (Oxf). 2010; 72(4):427– 435
3. Bianco SD, Kaiser UB. The genetic and mo-lecular basis of idiopathic hypogonado-tropic hypogonadism.Nat Rev Endocrinol. 2009;5(10):569 –576
4. Lalani SR, Hefner MA, Belmont JW, Daven-port SL. CHARGE syndrome. Available at:
w w w . n c b i . n l m . n i h . g o v / b o o k s h e l f / b r . fcgi?book⫽gene&part⫽charge. Accessed January 23, 2009
5. Verloes A. Updated diagnostic criteria for CHARGE syndrome: a proposal.Am J Med Genet A. 2005;133A(3):306 –308
6. Pinto G, Abadie V, Mesnage R, et al. CHARGE syndrome includes hypogonadotropic hy-pogonadism and abnormal olfactory bulb development.J Clin Endocrinol Metab. 2005; 90(10):5621–5626
7. Wheeler PG, Quigley CA, Sadeghi-Nejad A, Weaver DD. Hypogonadism and CHARGE as-sociation. Am J Med Genet. 2000;94(3): 228 –231
8. Vissers LE, van Ravenswaaij CM, Admiraal R, et al. Mutations in a new member of the
chromodomain gene family cause CHARGE syndrome.Nat Genet. 2004;36(9):955–957 9. Lalani SR, Safiullah AM, Fernbach SD, et al.
Spectrum ofCHD7mutations in 110 individ-uals with CHARGE syndrome and genotype-phenotype correlation.Am J Hum Genet. 2006;78(2):303–314
10. Jongmans MC, van Ravenswaaij-Arts CM, Pitteloud N, et al.CHD7 mutations in pa-tients initially diagnosed with Kallmann syndrome: the clinical overlap with CHARGE syndrome.Clin Genet. 2009;75(1):65–71 11. Kim HG, Kurth I, Lan F, et al. Mutations in
DOI: 10.1542/peds.2010-0164 originally published online November 1, 2010;
2010;126;e1594
Pediatrics
Milunsky
Andrew Dauber, Joel N. Hirschhorn, Jonathan Picker, Thomas A. Maher and Aubrey
Syndrome
Causing CHARGE
CHD7
Delayed Puberty Due to a Novel Mutation in
Services
Updated Information &
http://pediatrics.aappublications.org/content/126/6/e1594
including high resolution figures, can be found at:
References
http://pediatrics.aappublications.org/content/126/6/e1594#BIBL
This article cites 9 articles, 0 of which you can access for free at:
Subspecialty Collections
http://www.aappublications.org/cgi/collection/genetics_sub Genetics
following collection(s):
This article, along with others on similar topics, appears in the
Permissions & Licensing
http://www.aappublications.org/site/misc/Permissions.xhtml
in its entirety can be found online at:
Information about reproducing this article in parts (figures, tables) or
Reprints
http://www.aappublications.org/site/misc/reprints.xhtml
DOI: 10.1542/peds.2010-0164 originally published online November 1, 2010;
2010;126;e1594
Pediatrics
Milunsky
Andrew Dauber, Joel N. Hirschhorn, Jonathan Picker, Thomas A. Maher and Aubrey
http://pediatrics.aappublications.org/content/126/6/e1594
located on the World Wide Web at:
The online version of this article, along with updated information and services, is
http://pediatrics.aappublications.org/content/suppl/2010/11/05/peds.2010-0164.DC1
Data Supplement at:
by the American Academy of Pediatrics. All rights reserved. Print ISSN: 1073-0397.