Genome annotation for clinical genomic diagnostics: strengths and weaknesses
19
0
0
Full text
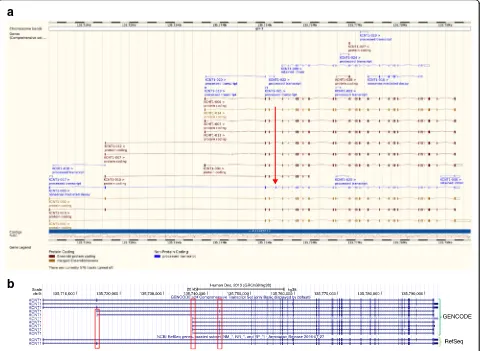
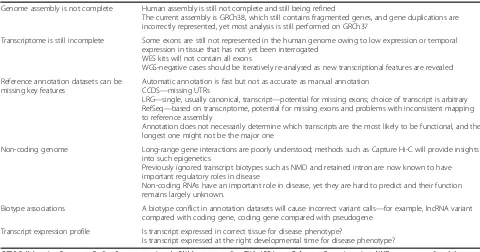
Figure
+7
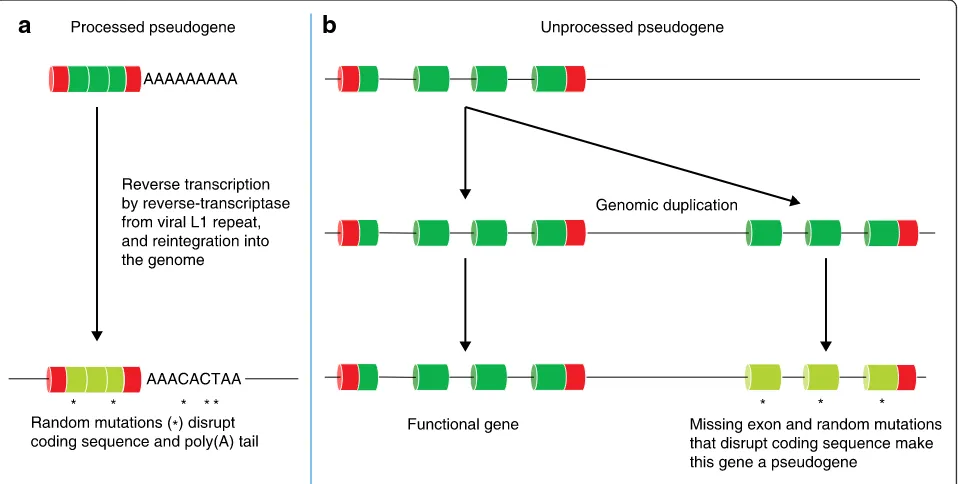
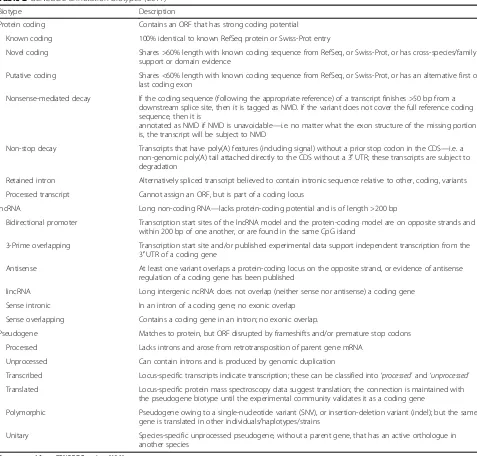
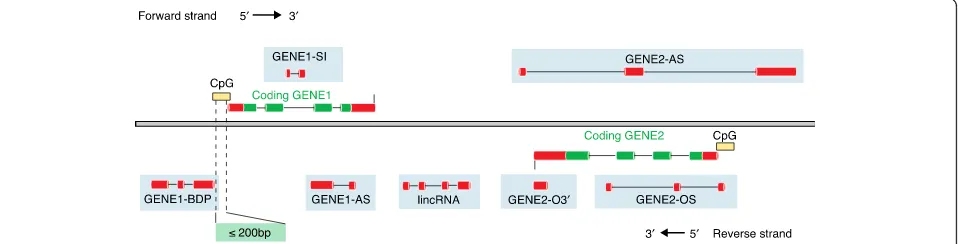
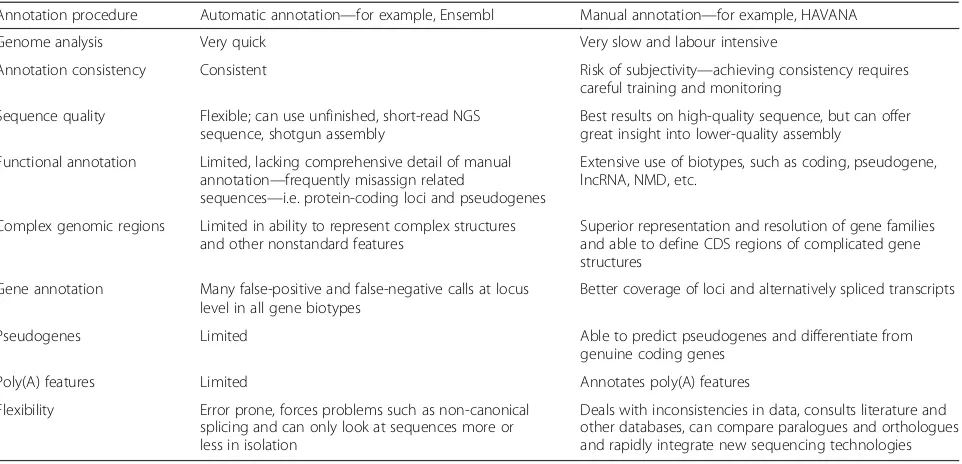
Related documents