All rights reserved INFORMATION TO ALL USERS
The q u ality of this re p ro d u ctio n is d e p e n d e n t upon the q u ality of the copy subm itted. In the unlikely e v e n t that the a u th o r did not send a c o m p le te m anuscript and there are missing pages, these will be n o te d . Also, if m ateria! had to be rem oved,
a n o te will in d ica te the deletion.
Published by ProQuest LLC (2017). C op yrig ht of the Dissertation is held by the Author. All rights reserved.
This work is protected against unauthorized copying under Title 17, United States C o d e M icroform Edition © ProQuest LLC.
ProQuest LLC.
789 East Eisenhower Parkway P.O. Box 1346
I m p r o v i n g P r o t e i n S t r u c t u r e P r e d i c t i o n
t h r o u g h D a t a P u r i f i c a t i o n
Fan Zhang (Mr.)
Submit in partial fulfilment for the
Degree of
Doctor o f Philosophy
From the
University o f Surrey
UniS
Department o f Computing;
School o f Electronics and Physical Sciences;
Division o f Chemistry;
School o f Biomedical and Molecular Science;
University o f Surrey
Guildford, Surrey GU2 5XH, UK
M arch 2007
Abstract
A ch iev in g the ability to p red ict the th ree dim ensional structure o f a p ro tein solely from its sequence is w idely ack n o w led g ed to be essential in tack lin g m ost genetic diseases. D eoxyribonucleic acids (D N A ) co nstitute chem ical blueprints fo r p ro tein construction, bu t once these b lu eprints are dam aged, the correct structure and function o f proteins, w h ich are constructed according to th ese b lueprints, can no longer be guaranteed. M o st genetic diseases are caused by irregularly constructed p ro tein s, and structural ab norm alities o f proteins m ay lead to irregular biological beh av io u r w ithin the hum an body, such as u nco ntrolled cell division. G enetic disease is one o f th e m ajor causes o f hum an death, from a v ery early age, and fo r centuries m ankind has tack led these diseases using a variety o f approaches. W ithin the research w orld this has becom e a w ide spread and international w ide activity, and the advancem ents in com m u nicatio n techniques and com putational hard w are m ake this research m o re exciting and effectiv e than it h as ever been.
It is w ell know n that p roteins are th e fo un dation o f hum an life, and th eir biological functions catalyze and control alm o st all the chem ical reactions w ith in the hum an body. T hese biological functions rely com p letely on the shape o f th e p ro teins in th ree- dim ensional space and th erefo re a k n o w led ge o f protein structure is required in advance to cure diseases caused by p rotein m alfunctions.
T he p roblem o f protein folding rem ains critical and as y et u n so lv ed in the literature. O w ing to the close correlation b etw een a p ro te in ’s structure and its biological functions, un derstanding three dim ensional protein structures becom es th e fro n t line o f th e w ar against protein disorder.
A lm o st all protein s autom atically fold into th eir unique shapes spo ntan eou sly w hen they are pu t into a m oist environm ent. T he fo ld in g process is to o fast and is im possible to be m onitored at the m om ent. C onversely , scientists determ ine the sequences and structures o f proteins sequentially, and exp lo re th e co rrelation betw een th e sequences and the structures. T hese discovered correlation s lead to the possibility o f co nd ucting a successful
prediction o f p rotein structure from seq uence alone. T he d isco very o f a successful m ethodology o f protein structure pred ictio n n o t only eno rm ou sly cuts dow n th e expense o f determ ining pro tein structure bu t also dram atically increases the efficiency. In general, i f successfully im plem ented, it w ill offer d rastic im provem ents in th e state o f th e art.
T here w ere outstand ing successes to w ards u n d erstan din g protein structures in the p ast few decades. X -R ay crystallograph y is one o f the favo urite tech niq ues used in determ ining pro tein structure (A bo la et al. 2 00 0;S m yth & M artin 2 0 00 ;Y affe 2005), and n uclear m agnetic resonance (N M R ) spectro scop y is an other w id ely used approach in screening structure o f proteins (H aw kes et al. 1980;M arkley 1989;V an, V, B ellon, & L anens 1988;W uthrich 1989). U sing th ese facilities to determ ine p ro tein structure used to be both tim e consum ing and reso urce intensive, and som etim es can go w ro ng if they w ere no t im plem ented correctly (N abu urs et al. 2006). T he invention o f h igh th ro u g h p u t screening techn o lo g ies granted the users the ability to screen ov er h u nd reds o f sam ples at th e sam e tim e and th erefo re enorm ou sly reduced the tim e co m p ared to the tim e co st o f sequential screening (C apelle, G urny, & A rv in te 2006), bu t unfortu nately the process is still expensive. A n altern ativ e ap proach to p rotein structure d eterm inatio n uses com putational algorithm s and m etho do lo gies to predict the structure o f a protein, based on applying m achine learning schem es to a set o f protein sam ples, w h o se structures are already resolved.
T he ultim ate targ et is to achieve th e ability to p red ict p ro tein structure based solely on its sequence. In o rd e r to fulfil this eventual p u rp ose, the p red ictio n o f p ro tein secondary structure is the first strategic step; a good secondary stru ctu re p red iction m ethodology p rovides a good starting p o in t to w ard tertiary structure p rediction. C hristian A n fin se n ’s sem inal discovery, in 1973 o f the d en atu ring process o f rib on uclease (A nfinsen 1973) laid th e foundation o f m od ern in silico research into p rotein structural determ in ation from sequence inform ation alone. In the p ast thirty years, various choices o f m achine learning schem es have been applied to the ap plicatio ns o f p ro tein structure p redictio n, h ow ever the reported pred ictio n accuracy rem ains less th an 80% .
In this thesis, th e au th o r pursues th e targ e t o f im proving accuracy o f protein structural p rediction through th e procedure o f data purification. A P rotein A ttribu tes M icro tun in g S ystem (P A M S ) is d ev eloped to p repare a variety o f new datasets as and w hen required.
F urtherm ore, a P rotein S tructural A ccu racy R eck on er (P S A R ) fram ew o rk is used to recom m end procedures th at m ight lead to hig h prediction accuracy. B y using the P SA R , it is show n th at using a refined d ataset generated by the P A M S, and im plem enting an appropriate w indow m echanism con siderab ly im proves the accuracy o f protein structure p rediction by 12% , giving a b est accuracy o f 90.97% . O n average, alm ost all classifiers th at are applied in the experim ents re su lt in accuracy increases o f 10% -15% . A list o f classifiers is categorized accord ing to th eir pred ictio n p erfo rm an ces and classification efficiencies. A few refined datasets are p ro posed as benchm ark datasets.
A part from the aforem entio ned achievem ents, ex am ination o f a total o f 3,135,393 p red ictions tasks, w hich carried out by the P S A R fram ew ork, yielded 139 ‘b e st’ and 73 ‘w o rst’ com binations o f am ino acid features descriptors. In this analysis, the ‘b e s t’ pred iction gave 82.34% , and the ‘w o rst’ pred ictio n gave 73.65% .
To achieve a g reater com putatio nal capacity the P S A R infrastru ctu re is hosted on the C ondo r (T hain, T annenbaum , & L ivny 2005) platform in the D ep artm ent o f C om puting, U niversity o f Surrey (G rid C om puting project, D epartm ent o f C o m puting, U niversity o f Surrey).
K ey w ords: P rotein S econdary S tructure P rediction; D ata P urification; H igh T h rou gh put C om puting; M achine L eaning.
Acknowledgments
F irst o f all I w ant to dedicate m y th ankful heart to m y parents, w ho nev er lose th eir faith in m e. To be honest, doing research in E ng lan d is nev er as easy as I im agined before I cam e to here. D urin g the p ast fo u r y ears, w h en ev er I h ad tro u b les th ey com forted m e; w h en ev er I w as lonely they show ed th eir generous love to m e, en couraged m e to p lace fo otsteps firm ly to w ard th e targ e t o f finishing th is research.
P rofessor D avid P ovey (P rofesso r o f C o m p u ter A ided C hem istry) is m y hero. Fie helped m e in obtaining an oppo rtun ity to w o rk in th is fascinating research area. H e provided m e easy access to eq uip m ents and placed m e in C ecil D avies L abo ratory, D ivision o f C hem istry. D uring m y tim e been there, I acqu ired the necessary kno w ledg e from a chem ical view point to fo rm this thesis. P ro fesso r Povey, as m y secondary supervisor, inspired m e to develop the infrastructu re and k ept encouraging m e to delve d eep er in to this research area. O bviously the saying “he is the hero o f the C hem istry D ivision ” is very true.
I also o ffer m y th anks to P ro fesso r P aul K rau se (P ro fesso r o f S oftw are E n gineering) for offering m e an extrao rdinary ch ance after I had difficulties. A s m y principal supervisor, his un bounded kn ow ledge in softw are eng in eerin g inspired m e in v ariou s aspects to form the P A M S /P S A R fram ew ork. H is kindn ess and gen ero sity is so im portant fo r the exciting and relaxing w o rk en vironm ent in th is p ro ject and bro ugh t lots o f fun.
T hanks to D r Ian H am erton fo r p ro viding ex cellen t p ro o f read in g o f the com position o f this thesis. T he tim e th at I spen t in th e roots cafe in the p ast th ree years w ith D r B rendan H ow lin, D r Ian H am erton, and D r G abriel C avalli-P etrag lia w as v ery pleasant. T he friendly environm ent th at w e created w as delightful and k ept the p ro ject g oing w ell.
M r C hris B radshaw , as the IT supp ort m an ag er in School o f B io m ed ical and M olecu lar Sciences in U niversity o f S urrey p ro v id ed lots o f facilities for this project. O din.ch em .su rrey.ac.u k, as the m ajo r server fo r this p roject w as fully supported by C hris.
I w o uld like also th an k m y friends V icto r Z hou (E lectronic E n gineering@ S urrey), Jin W u (C om puting@ L eeds), Paul T ap p er (C om puting@ B ath), and Joh n E llio tt for keeping m e com pany w hile I stayed in G uildford.
Contents
Contents
A b s tra c t...ii A c k n o w led g m en ts...v C o n te n ts ... vii L ist o f F ig u re s ...xi L ist o f T a b le s ... xiii L ist o f T e rm in o lo g y ...xiv 1 In tro d u c tio n ...1 1.1 M edication in H um an C ivilization...11.2 Proteins and M odern Structural B io lo g y ... 2
1.3 Structural B ioinform atics... 3
1.4 Statem ent o f H y p o th esis...4
1.5 Structure o f the T h e s is ... 5
2 P rotein and P rotein S tru c tu re ...6
2.1 Protein is L ife...6
2.2 The Physical C om position o f P ro tein s... 7
2.3 The Process o f Protein F o ld in g ... 10
2.4 Protein C om position and S tru c tu re s... 11
2.4.1 Am ino A cids as B uilding Blocks o f P ro te in ... 12
2.4.2 Prim ary Structure as Sequence Indicator... 13
2.4.3 Secondary Structure as Pattern o f General 3D Form ... 14
2.4.4 Super-Secondary S tructure... 17
2.4.5 Tertiary S tructure...17
2.4.6 Quaternary S tru c tu re ...19
2.5 Essentiality o f Protein S tru c tu re ... 19
2.6 C onclusions... 19
3 P ro tein Secondary S tructure P re d ic tio n ... 21
3.1 O verview o f General Protein Structure P re d ic tio n ...21
3.2 H istorical View o f Protein Secondary Structure P re d ic tio n ... 23
Contents
3.2.1 M ethods o f First G eneration... 23
3.2.2 M ethods o f Second G e n e ra tio n ...24
3.2.3 M ethods o f Third G en e ra tio n ... 25
3.3 Review o f Prediction M ethods... 26
3.3.1 Chou-Fasm an M e th o d ...26
3.3.2 G O R M e th o d ... 27
3.3.3 Q ian-Sejnow ski M e th o d ... 27
3.3.4 A pplications U sing Support Vector M achine... 28
3.3.5 M ethods U sing B ayesian N etw ork C lassifiers... 30
3.4 The M easurem ent o f Prediction A ccuracy... 32
3.4.1 Per-residue Three-State A ccu rac y ... 32
3.4.2 Per-Segm ent O verlap A ccuracy (S O V )... 33
3.5 Cross-V alidation and Confusion M a trix ...34
3.6 C o n c lu sio n ...35
4 P rotein A ttributes M icrotu ning S y s te m ... 36
4.1 General Introduction o f the P A M S ... 37
4.2 Characteristics o f the P A M S ...38
4.2.1 Reliability o f the P A M S ...39
4.2.2 C om patibility o f the P A M S ...40
4.2.3 Usability o f the P A M S ... 40
4.3 System A nalysis and D esign o f the P A M S ... 40
4.3.1 The D evelopm ent Platform o f P A M S ... 41
4.3.2 The M odern Techniques that PAM S has Im plem ented...43
4.3.3 The Biological Resources That the PAM S Flas A d o p ted ... 44
4.3.4 The M achine L earning Classifiers Package that the PAM S has A pplied...46
4.4 The G raphical U ser Interface o f the P A M S ... 50
4.5 The Introduction to M odules’ F u n ctio n ality ... 56
4.5.1 The DB H andler M anages the D atabase... 57
4.5.2 The W indow Fram e M odule Turns Sequences into Segm ents... 57
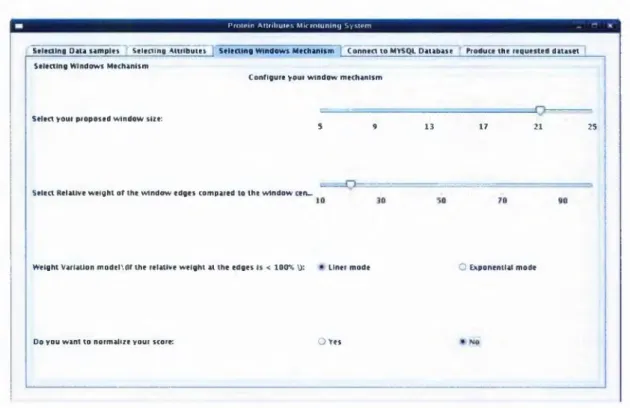
4.5.2.1 D eciding an Optimal W indow L e n g th ... 58
4.5.2.2 D eterm ining the W indow W eight Variation M odel... 59
4.5.3 The Feature Calculus Com putes the A ttribute Value for the C en tral...62
4.5.4 The A RFF W riter O utputs the Datasets in ARFF F o rm at... 63
4.6 D ealing w ith the M issing V alue...64
4.7 C o n c lu sio n ... 64
Contents
5 P rotein Structural A ccuracy R eck o n e r...65
5.1 M otivation behind the PSA R D evelopm ent... 65
5.2 Characteristics o f the PSA R Infrastructure...66
5.3 Functional M odules o f the PSA R Infrastructure... 68
5.3.1 B ioD ata-O riented M o d u le s...69
5.3.1.1 The D atabase... 69
5.3.1.2 The P A M S ...70
5.3.1.3 The C la ssifie rs...72
5.3.2 C onfiguration-O riented M o d u le s... 72
5.3.2.1 Feature Sam pling M o d u le... 72
5.3.2.2 Protein Sam pling M o d u le ... 73
5.3.2.3 W indow Sam pling M o d u le...74
5.3.2.4 C om bination Sam pling M o d u le...75
5.3.3 Result-O riented M odules...75
5.3.3.1 D atabase m o d u le... 75
5.3.3.2 Results C apturer M o d u le ... 77
5.3.3.3 Results R eview ing M o d u le ...77
5.4 Safety Evaluation o f the PA M S/PSA R Fram ew ork...77
5.4.1 System Security o f the Hosts w ith the PA M S/PSA R In sta lle d ... 78
5.4.2 Reliability o f the Procedures D efined by P A M S /P S A R ... 80
5.4.3 Validity o f the Biological D a ta se t... 80
5.5 Perform ances E valuation o f P A M S /P S A R ...80
5.6 C onclusions... 81
6 Im plem entation o f the P S A R F ram ew o rk ...82
6.1 Sam pling the Feature D escrip to rs... 82
6.2 D eterm ining the N um bers o f A pplied Feature D escrip to rs...86
6.3 Ranking the 3080 Protein S a m p les...88
6.4 Selecting W indow L ength...92
6.5 Selecting L inear or E xponential... 102
6.6 Selecting Com binations o f A m ino A cid Feature D escriptors... 106
6.7 C onclusions... 109
7 F uture W o rk ...110
7.1 Technical Im provem ents for P S A R ...110
7.2 Scientific Im provem ents for P S A R ...111
7.3 From Secondary Structure to Tertiary Structure...112
Contents
7.4 General C onclusion... 113
List o f Figures
List of Figures
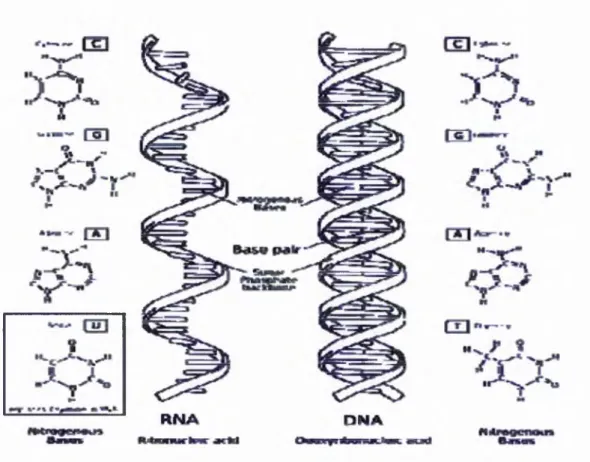
Figure 2-1: DNA - RN A - PROTEIN (© the Nobel Foundation) ... 7
Figure 2-2: Universal Genecode (©Utah University) ... 7
Figure 2-3: Comparisonsbetween DNA andsinglestranded RNA. (GNU Licensed) ... 9
Figure 2-4: Structureofaminoacid (showingzwitterionicstructure) ...12
Figure 2-5: Structureofalanine (showingzwitterionicstructure) ...12
Figure 2-6: Structureofglycine (showingzwitterionicstructure) ...12
Figure 2-7: A tripeptidewithtwopeptidebonds (showingzwitterionicstructure) ... 13
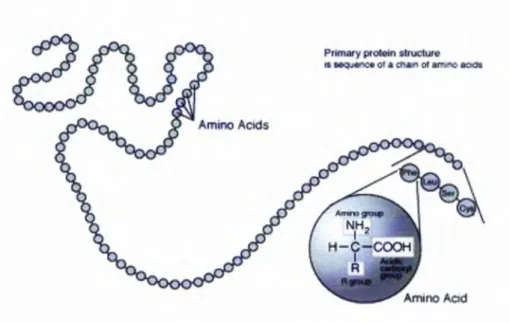
Figure 2-8: Proteinprimarystructure. (GNU Licensed) ...14
Figure 2-9: Threedimensionalstructureofthemyoglobinprotein. (GNU Licensed) ... 16
Figure 2-10: Stickrepresentationofoverallstructureofinsulin... 18
Figure 3-1: Proteinstructurepredictionflowchart (by Rob Russell, EMBL)... 21
Figure 3-2: Architectureof BAYESPROT (Chinnasamy, Sung, & Mittal 2004)... 31
Figure 3-3: TAN Bayesian Classifier (Chinnasamy, Sung, & Mittal 2004)... 31
Figure 4-1: Structureofthe PAMS... 37
Figure 4-2: Thesampleofprotein: 2i 1 bwastakenfromthe RS 126 dataset... 45
Figure 4-3: Thesampleofprotein: Ibncwastakenfromthe CB396 dataset... 45
Figure 4-4: ARFF headerinformationsection, partone. (Professor David Poveynow)...48
Figure 4-5: ARFF headerinformationsection, parttwo... 48
Figure 4-6: ARFF datasection... 49
Figure 4-7: The PAMS, datasetselectionpage... 50
Figure 4-8: The PAMS, attributesselection 1... 51
Figure 4-9: The PAMS, attributesselection, II...52
Figure 4-10: The PAMS, configuringthewindowmechanism...54
Figure 4-11: The PAMS, MySQL databaseconnection...55
Figure 4-12: The PAMS, generatingthetargetdatasets... 56
Figure 4-13: A windowframe, withthelengthof 11...57
Figure 4-14: Slidingthewindowonepositiontotheright... 58
Figure 4-15: A windowframewithweightsassigned... 59
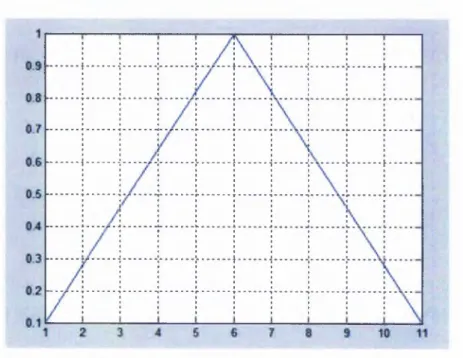
Figure 4-16: Linearweightvariationmethodwhen N = 11...60
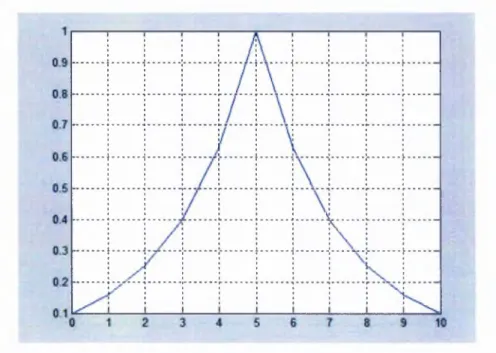
Figure 4-17: Exponentialweightvariationmethodwhen N = 11...61
Figure 4-18: Thediverseweightassigningmethod... 62
Figure 4-19: Weighedwindowframe... 63
Figure 5-1: distributed PSAR infrastructure... 67
Figure 5-2: Infrastructureofthe PSAR...68
Figure 5-3: Nessusclientforsecurityvulnerabilitiesassessment...78
Figure 5-4: NMAP reportingthesecurityassessments...79 xi
List o f Figures
Figure 6-1: PSAR terminaloutputforattributesampling...83
Figure 6-2: PSAR terminaloutputforexperimentofnumbersofappliedfeaturedescriptors. . 87
Figure 6-3: Predictionvariesalongwithnumbersofappliedfeaturedescriptors ...88
Figure 6-4: PSAR terminaloutputofproteinsampling...89
Figure 6-5: Thequalityofproteinchains... 91
Figure 6-6: Terminaloutputsofselectingwindowlengthwith FAN + exponentialmodel; 93
Figure 6-7: Selectingwindowlengthby FAN + Exp... 94
Figure 6-8: PSAR terminaloutputandplotshowingselectingwindowlength (IBk + Linear). . 95
Figure 6-9: PSAR terminaloutputandplotshowingselectingwindowlength (Kstar + Exp). .. 96
Figure 6-10: PSAR terminaloutputandplotshowingselectingwindowlength (LMT + Exp). ... 97
Figure 6-11: PSAR terminaloutputandplotshowingselectingwindowlength (SMO + Exp).... 98
Figure 6-12: PSAR terminaloutputandplotshowingselectingwindowlength (Jrip + Exp) 99
Figure 6-13: PSAR terminaloutputandplotshowingselectingwindowlength (PART + Exp). 100
Figure 6-14: Windowlengthinfluencestopredictionresultwithdifferentclassifiers 101
Figure 6-15: Resultsthatobtainedwhenclassifiersworkedwithlinearweightvariation
MODEL... 103
Figure 6-16: Resultsthatobtainedwhenclassifiersworkedwithexponentialweightvariation
MODEL... 104
Figure 6-17: Performancesofclassifierswithlinearvariationmodel... 105
Figure 6-18: Performancesofclassifierswithexponentialvariationmodel... 105
Figure 6-19: Combinationsoffeaturedescriptorsproducedpredictionaccuracygreaterthan
84%... 107
Figure 6-20: Occurrencesoffeatureswithpredictionaccuracies >84%... 107
Figure 6-21: Combinationsoffeaturedescriptorsproducedpredictionaccuracylessthan 73%.
108
Figure 6-22: Occurrencesofattributeswithpredictionaccuracies <73%... 108
Figure 7-1: PSAR Interface Design...111
List o f Tables
List o f Tables
Table 2-1: Listof Amino Acids (GNU Licensed) ... 9
Table 4-1: Theweightvaluesthatassignedbylinearweightvariationmodel...60
Table 4-2: Theweightvaluesthatassignedbyexponentialweightvariationmodel...61
Table 5-1: Theconfigurationofaparticulardataset...71
Table 5-2: Configurationoffeaturesamplingmodule... 73
Table 5-3: Configurationforproteinsamplingmodule... 74
Table 5-4: Configurationforwindowsamplingmodule... 74
Table 5-5: Sectiondefinitioninthetable, whichrecordsresults...76
Table 6-1: Toptenrankedattributesinexperiment 3.3.1 byusing Bayes .TAN... 84
Table 6-2: Toptenrankedattributesinexperiment 3.3.1, byusing IB 1... 85
Table 6-3 : Classificationoftop 168 rankedattributesfor Bayes.TAN... 85
Table 6-4: Thepatternofselftestingranks... 90
Table 6-5 : Thepatternofpeptidechainlength...90
Table 6-6: Performancesoflearningschemeswithweightvariationmodel...102
List o f Terminology
List o f T erm inology
Protein:
Proteins are large organic com po un ds m ade by com b inatio ns o f th e tw enty natural o ccurring am ino acids. P ro teins are essential fo r living life and are a m ajor com position o f living cells.
A m ino A cid:
A m ino acids are any m o lecu les th a t co ntain both am ine group and carboxyl functional groups. A ll am ino acids share a com m on structure w ith a central a
-carbon. In th e thesis am ino acids co nstructs blocks o f proteins.
D eoxyribonucleic A cid (D N A ):
D N A is a nucleic acid th a t contains the g enetic in structions fo r biological developm ent o f a cellu lar form o f life or a virus. D N A co ntains tw o com plem entary strands, w hich tw ist to g eth er and form a double helical shape. Tw o com plem ents o f helices in the pro cess o f D N A rep lication , before the cell holding it divides to form tw o d au g h ter cells.
R ibonucleic A cid (R N A ):
R N A is a n ucleic acid th at co ntains ribose rings and uracil, unlike D N A . It is transcribed from D N A by enzy m es called R N A p o lym erases and fu rther processed by o ther enzym es. R N A serves as the tem p late fo r translatio n o f genes into proteins, tran sferrin g am ino acids to the ribosom e to form proteins.
Protein A ttributes M icro tu n in g S ystem (P A M S ):
T he P A M S is th e softw are th at w as develop ed in this research to g enerate training and testing dataset auto m atically from unform atted bio lo gical d ata resources. T he invention o f the P A M S allo w ed the pro ced ures o f data p re-p rocessing , data cleansing, data discretizatio n etc, to be autom ated.
List o f Terminology
P rotein Structural A ccuracies R eck o n er (P S A R ):
T he P S A R in frastructure w as d evelop ed in order to p erfo rm a d esign ated series o f prediction task s au tom atically according to th e specification s subm itted by the users. T he P S A R infrastructure is a distributed com pu tation al platform th at requires m inim um hum an intervention.
Chapter 1: Introduction
1 Introduction
1.1 M e d ic a tio n in H u m a n C iv iliz a tio n
M odern archaeological research has established th at the earliest hu m an civilization dates back to som e 7,000 y ears (i.e. 5000 B .C .). T he know led ge o f m ed ication has passed throu gh m any generations, d espite the rise or fall o f a particu lar dynasty. E arly treatm ents m ostly focused on p e o p le ’s beliefs and o rig in ated from a variety o f sources such as m yth, religion and experiences in using herbs (B eren zon & S aavedra 200 2;C ow ley, Y oung, & R affin 1992;G ross 1992;K arpoziios & Pavlidis 20 04 ;P arry jo n es & P arryjones 1991;Peponi 2002;R am outsaki et al. 2001). F o r exam ple th e trad itional C hinese herbal m edicine cam e from th e m yth o f a brav e do cto r w ho tasted all typ es o f traditional herbal m edicines and w ro te the “bo o k o f St N o n g ” . T his precious bo ok is still heavily used in m od ern traditional herbal m edicine research to th is day. T hese ty p es o f m edication records are no t only excav ated in C hina, b u t also in o ther an cien t civilization s, such as E gypt and ancient B abylon (A ndersen 1997;M anyam 1992;M artin-A raguz et al. 2 0 0 2 ;0 c k litz 1997;T revisanato 2005). T he papyri dug ou t in E gypt, k n ow n as the “Edw in S m ith” and “ G eorge E bers” scrolls, repo rted com plicated treatm ents for cancer. The Sm ith papyrus describes surgery, w hile the E b er p apy rus outlines pharm aco log ical, m echanical, and m agical treatm ents. H um ank in d never gives up try in g to d efeat diseases and attem pting to extend life. •
In m o d em science, along w ith the advan ces in the u n d erstan din g o f hum an anatom y, som e o f the m ysterious d iseases have been exam in ed using m o d em th eorem s and tech nologies. F or instance, the use o f th e m o d em m icroscope p ro vides oppo rtu nities to study diseases and th eir functions such as th e H IV virus, and cancer cells, w hile cellu lar p ath ology provides th e scientific basis fo r the m o d em p ath o lo g ic study o f these diseases. T h e w ord “ can cer” cam e from the L atin w ord expression “ carcin o m a” , defined by the great do cto r H ippocrates (C raik 2000). T he w ord itse lf reflects the observation th at the dissected surface o f a solid m alig n an t tu m o u r looked like a crab, w hich in G reek is denoted as “ carcinos” . A part from can cers th ere are m ore th an 20 diseases th at have been
/
identified as being caused by p ro tein m isfo ld ing , th erefore th ey are called “p rotein m isfolding d iseases” (E llisdon & B ottom ley 2004).
1.2 P r o te in s a n d M o d e r n S tr u c tu r a l B io lo g y
P roteins carry o u t essential biolo gical fun ctio ns in every cell o f living organism s. To carry out these functions correctly, p ro tein s have to fold th em selv es into th eir unique favoured three dim ensio nal shapes in a m o ist environm ent. T he p ro cess o f p rotein folding is rem arkably quick, ho w ev er som etim es it can go w rong. O nly in the 1990s did it becom e clear th at w rongly fo ld ed p ro tein s are involved in th e d evelo pm ent o f m any diseases, such as am yloidoses (B u x b au m & T ago e 20 00 ;C oh en 1994;Falk & S kinner 2000), A lzh e im e r’s disease (L age 2006), prio n diseases (F errer 2 001 ;K o gan et al. 2002), and som e cancers. Im provem ents in the un derstan d in g o f p rotein structures dram atically changed the w orld o f new drug discov ery and becam e th e focus o f p harm aceutical research and b iotech com panies; the future o f genetic th erapy is very prom ising.
In the p ast few decades, tech niqu es fo r identifying protein sequences from the corresponding gene b lueprints hav e becom e increasingly m ature and efficient. H ow ever, even w ith the success o f high th ro u g h p u t screening ap plication s, the pro cess o f d eterm ining p rotein structure ex perim en tally is still expensive and tim e consum ing. T hus, structural d eterm in ation ten d s to outpace sequence d eterm ination leading to an increasing gap betw een them . C om putational b iologists pursue th e targ e t o f determ in in g protein structure according to sequ ence alone, try in g to avoid the enorm ous expense o f d eterm ining p rotein structures experim entally , by using X -ray cry stallog raphy o r nu clear m agnetic R esonance (N M R ) spectroscopy.
A lthou gh the ac h iev em en t o f exp erim ental d eterm ination o f th e structure o f biological m acrom olecules is outstanding, it still ca n n o t m eet th e cu rren t dem ands, w hich grow every day. C om putational b io logists are dev elo p in g theorem s and m etho do lo gies to p red ict the p rotein structure in siiico, and in the p ast few decades, have achieved rem arkable progress.
_________________________________________________________________ Chapter 1: Introduction
Chapter 1: Introduction
1.3 S tr u c tu r a l B io in fo r m a tic s
In the early 1960s C hristian A n fm sen and his co lleagu es investigated the p ro tein rib onuclease, and carried ou t the exp erim en t w hose results w ere co nsid ered to be the corn erstone o f m odern pro teo m ic science (A nfin sen 1973). T he ribo n u clease contains 124 am ino acids, and folds into a //-sh eet and th ree a-helices. T he structure o f ribo nu clease is stabilized by several d isulphide b rid g es and m ay be collapsed by add in g certain chem icals, o r sim ply by the application o f heat. T he process o f break in g th e stable structure o f p rotein is called “den atu rin g ” , and by break ing th e disulp hid e bon ds th e active structure o f ribonuclease turns into a useless ball. T he process o f denaturing ribo nu clease can be reversed by the restoration o f the en vironm ent, m ore precisely by rem oval o f the previously added chem icals, o r by low ering th e tem p erature to the origin status. T he ribonuclease then folds b ack into its natural functional state on its ow n. From the ex perim ent A nfinsen et, al, co n clu d ed th at the am ino acid seq uence determ ined the shape o f the protein, a resu lt w h ich b ro u g h t A nfm sen the N obel P rize in C hem istry in 1972.
A n fin se n ’s discovery show s th a t it is p ossible th at protein stru ctu re can be deduced by applying certain folding fu nctions in its sequence. The ‘trial and erro r’ m ethod w as proved to be infeasible by C yrus L evin th al in 1966, and w as nam ed as the “L evinthal P arad o x ” (L E V IN T H A .C 1968). L evinthal stated th at if o ne con sid ered a sm all protein, p erhaps com prising 100 am ino acid residues, and allocated each flexible residu e to ad op t only tw o different spatial con form atio ns. T hen theo retically it w o u ld still take 100 billion years to try all possib ilities. T here are several o ther appro aches to w ard bypassing the L evinthal P aradox, stated in o ther p u blication s (F inkel'shtein & B adretdinov 1997;H onig
1999;K arplus 1997).
T here are a few m ethodologies availab le in the literature to p red ict p ro tein tertiary structure com putationally (C hiu & G oldstein 2 0 00 ;F etrow & B ry ant 1993). T o p red ict th e tertiary structure o f a protein, it is good to have a good accurate secondary structure prediction m ethod in hand to p rovide assistance (E yrich et al. 1999). D esp ite p ro ducing novel m achine learning algorithm s, th is thesis discusses th e po ssib ility o f im proving protein secondary structure pred ictio n from prop osing p ro ced ures th at purify biological data.
Chapter 1: Introduction
C urrently in the literature, com parative m od ellin g is the m o st co m m o nly studied m ethod o f protein secondary structure p redictio n. It has achieved good records in p redicting the secondary structures o f proteins, w h ich share sequence identities, but is n ot necessarily good in predicting p roteins, w ith less sequence sim ilarity (less than 25 % sequence identity). P rotein th read in g tech n iq u es w ere d eveloped to cope w ith the situation th at pro tein s share no significan t sequence identity, but share a certain degree o f conform ational sim ilarities. T he thesis m akes no use o f hom olog y inform ation, y et still achiev es accurate results. T he P rotein A ttrib utes M icrotun ing S ystem (P A M S ) softw are packag e presen ted in th is th esis p ro vides a co nv enient w ay to prep are th e train in g dataset and testing dataset; and the p ro d u ced d atasets are evaluated w ithin th e P rotein S tructural A ccuracy R eck o n er S ystem (P S A R ) infrastructure. T he m eth od olog y proposed in this ex perim ent is a statistical, tem plate-free appro ach to th e p rob lem o f p ro tein secondary structure prediction. T he P A M S /P S A R fram ew o rk can perfo rm v ario us tasks aim ed at eith er obtaining good pred ictio n accuracy o r fo r discovering p attern s in biological data.
In general, the P A M S /P S A R infrastru ctu re prop osed in th is th esis fulfils several intended tasks. F irst o f all, the h ypo thesis o f this thesis is verified ; seco nd ly, certain m achine learning classifiers are disco v ered to p red ict p ro tein secondary structure m ore accu rately th an others; at the sam e tim e a few o th ers are d isco vered to perform the p redictio n efficiently. T he F D 232 dataset is prop osed as a good p ro tein can did ates set fo r p ro tein secondary structure prediction. In general, th e P A M S /P S A R infrastructure offers an insight into the correlations rev ealed by the pu rity o f biological sam ples and the accuracy o f p rotein structural prediction.
1.4 S ta te m e n t o f H y p o th e s is
“A s th e p u rity o f dataset is re fin e d a n d th e “quality ” is improved, th e classification o f dataset becom es better a n d therefore th e prediction accuracy will im prove regardless o f w hich classifier is being used.”
Chapter 1: Introduction
1.5 S tr u c tu r e o f th e T h e sis
T he structure o f this th esis is assem bled as follow ing: C hap ter O ne introduces th e fundam ental concepts and gives a h isto rical review o f the state o f th e art; it also contain s the hypothesis. T he second ch ap ter rev iew s p ro tein and p ro tein structure from a biological view point, and indicates the con cepts o f prim ary , secondary, tertiary, and q uaternary protein structure. C hap ter T hree review s th e state o f th e art o f p ro tein secondary structure prediction, and the three generatio ns o f protein secondary structure predictio n m ethodologies are identified and studied. C hap ter F o ur describes th e develo p m en t o f the PA M S. C hapter F ive p resen ts th e develo p m en t o f the P S A R infrastru cture, along w ith the evaluation o f it. In C hap ter Six the exp erim en tal results o b tain ed by im plem enting the P S A R in frastructure are illustrated. F inally in C hap ter Seven, fu rth er w o rk and possib le extensions o f the current softw are p ackag es and softw are fram ew o rk are described.
Chapter 2: Protein and Protein Structure
2 Protein and Protein Structure
2.1 P r o te in is L ife
T he m o st appropriate phrase to d escribe th e co ncep t o f p rotein from the biological v iew p o in t is “p ro tein is life” . P rotein is essential to the structures and fun ction alities o f living cells, and it is su ggested th at p ro tein form s nearly 75% o f th e dry w eig h t in the h um an body. P roteins are one o f the fou nd atio ns o f carb on -based life form s living on the E arth, they are com plicated, high -m o lecu lar-m ass organic com pounds, m ade up from 20 naturally occurring am ino acids jo in e d to g eth er by p eptid e bonds. W ith the u nlim ited com bin ations o f these tw enty b u ild in g blocks, m illions o f proteins are form ed in nature, and there are m ore o r less 100,000 pro tein s w ithin the hum an body, carrying out various functions.
P roteins such as enzym es cataly ze ch em ical reactions w ith in the hum an body, such as hy dration o f a carbon dioxide (P ihar 1965); or m odification o f sm all organic m olecules. E nzym es are involved in the pro cess o f read in g genetic inform ation from th e D N A , th e first pro cedures o f proteins synthesis. P roteins are fun ctio ning as transpo rters w ithin the hum an body, carrying nutrien ts and o th er relev an t m olecules around all ov er the body thro u g h o u t the cerebral circu latio n lym phatic system (B eav en & M cE w an 1969;H A L L G R E N 1954). P roteins such as collag en contain th ree po ly pep tide strains, each o f w h ich is a left-handed a-h elix . T h e th ree h a lf helix strands tw ist to g eth er by num erous hydrogen bonds, to form a stron g stable physical structure. T hese bundles o f proteins are the m ajo r com p o n en t o f con nective tissue in living anim als, also giving external cellu lar structure (B unyaratavej & W ang 2 00 1;Jo nes 1976;L achm an et al. 1992;L ow ther 1978). T ransm em brane p ro tein s form pores in cellu lar m em branes allow s ions to pass through (F leishm an, U nger, & B en-T al 2006). T he transp ortatio n o f proteins relies on som e o ther p ro tein s as w ell. P ro teins are critical in th e pro cess o f the im m une system fighting again st the intrusion o f harm ful bacteria and viruses. T he effectiven ess o f the im m une system h ig hly depends on th e p ro ductio n o f antibo dies w h ich are proteins capable o f b inding to specific fo reig n p articles such as b acteria and viruses, disabling the
Chapter 2: Protein and Protein Structure
harm ful effects (C H E R C H E N K O 1963;K osm as, L inardou, & E penetos 1993;Scallon et al. 2 006;T uorm aa 1988). In short pro teins are the foundation o f life.
2 .2 T h e P h y s ic a l C o m p o s itio n o f P r o te in s
Proteins are constructed by v ariou s ty pes o f R N A s w ithin cells. T he DN A carries the inform ation o f inheritance like a giant book o f blueprints, w hich instructs the construction o f biological m olecules w ithin living organism s. A ny m istakes occurred in the gene code have the potentials to cause disastro us conseq uen ces - genetic d iso rd ers (C ou rn oy er et al.
1990;K ingston 1989;M ayeux 2 0 0 5 ;P o n z d e 1994). DNA Replication RNA Transcription RNAPotyi RNA Processing Messenger RNA^' ’rotein Translation C E LL N U C LE U S Chromosome Nuclear membrane
Universal Genetic Code
(DNA format) T T - Phe \ CCC” CTA-CAT-Hw 'S) W CAA-Gin J CAG-Glnl GGT-Gty) GGC-Gly) GGG-Gly
F igure 2-1: DNA - RNA - PR O TE IN (© the N obel Figure 2-2: U niversal G ene code (© U tah U niversity) F oundation)
Chapter 2: P r o te in a n d P r o te in S tr u c tu re
A s represented in the above d iagram Figure 2-1, D N A records the bu ild ing blu eprin t o f a p articu lar protein. T he m essen g er R N A (m R N A ) transpo rts a p iece o f gene code ou t from the cell nucleus, and tran sp o rt R N A (tR N A ) brings building b lo cks - am ino acids to form the p rotein p o lypeptide chains. A ll g en etic cod e is presented by a set o f four, e.g. A, D , T an d G. E ach am ino acid is encoded by three codons, as p resented in Figure 2-2. A D N A gene code “ G C T T G C G A T G A A ” m ay p re sen t a sequence o f am ino acids as “A C D E ” . A p a rt from the code o f am ino acids, several codons also indicate ex tra inform ation bits such as the start indicator and end p o in t o f a protein. In the follow tab le a list o f acronym s o f am ino acids is presented:
A m ino Acid 3-L e tte r 1-L e tte r S ide ch ain p o la rity
S ide ch ain ac id ity o r b asicity
H y d ro p a th y index
A lanine A la A nonpolar neutral 1.8
A rginine Arg R polar strongly basic -4.5
A sparagine Asn N polar neutral -3.5
A spartic acid Asp D polar acidic -3.5
Cysteine Cys C polar neutral 2.5
G lutam ic acid Glu E polar acidic -3.5
G lutam ine Gin Q polar neutral -3.5
Glycine Gly G nonpolar neutral -0.4
H istidine His H polar weakly basic -3.2
Isoleucine lie I nonpolar neutral 4.5
Leucine Leu L nonpolar neutral 3.8
Lysine Lys IC polar basic -3.9
M ethionine M et M nonpolar neutral 1.9
Phenylalanine Phe F nonpolar neutral 2 .8
Proline Pro P nonpolar neutral
Chapter 2: P ro te in a n d P ro te in S tr u c tu re
Threonine Thr T polar neutral -0.7
Tryptophan Trp W nonpolar neutral -0.9
Tyrosine Tyr Y polar neutral -1.3
Valine Val V nonpolar neutral 4.2
Table 2-1 : List of Amino Acids (GNU Licensed)
T he am ino acids are bonded to g eth er by d ehydrating a w ater m olecule from the am ino group and carboxyl group to form a peptide bond w hich is quite strong. O nce the bond has been form ed, it requires large am o un t o f energy (vary from lOOkJmol' 1 to 3 0 0 k J m o r’, depends on the types o f am ino acid s to form the peptide bond.) to be broken. The follow ing diagram Figure 2-3 represents the com parison o f the DN A structures on a double helix and the single stranded R N A structure:
Chapter 2: P r o te in a n d P r o te in S tr u c tu re
T he proteins perform th eir desig n ated fu nctio ns after they are form ed w ith in living cells. T he cell can regulate the p resence o f a certain p rotein by m arking unw anted p ro tein s w ith a label consisting o f the polypep tide u biquitin. L abelled p ro tein s are th en broken dow n, or degraded rapidly in cellular “w aste d isp o sers” called proteasom es (C iech an ov er 2005;C iech an o v er 2006). T he re leased am ino acids can be reused again to form an other protein.
2.3 T h e P r o c e s s o f P r o te in F o ld in g
P roteins tend to fold into th e ir favourite three dim ensional shapes in a m o ist environm ent. T he process o f folding is driven by in trinsic p hysical forces, w h ich appears u nclear in the literature. T here are several statistical analyses to this problem , p ro ducin g m atrices, th a t m odel inter-residue co ntact energy b etw een d ifferen t ty p es o f am in o acids (O liv eberg et al. 1998;W ang & L ee 2000). T he tim e co st o f p ro tein fo ld in g varies from m icro seco nd s to nanoseconds, dependin g on the size and com plexity o f it (B aldw in 1994;C han 1998;ICubelka, H ofrichter, & E aton 2 00 4;M cC am m on 1996). H o w ever, ob serving the process o f protein folding in real tim e is n o t a possible task at the m om ent, even w ith the m ost accurate m icroscope and m ost sophisticated cam era. T he folded p rotein is flo ating in a liquid environm ent and looks like a tiny crystal subm arine u nd er w ater.
P roteins denature i f the stable “p ea cefu l” en viron m en t goes m ad - in a d enaturin g c o n d itio n 1. B iologically say, th e den atu red proteins are useless m ass balls, no t capable to carry out th eir biological functions. T he pro cess o f h eatin g th e solven t dam ages th e protein : as the tem perature increases th e protein w ithin gains energy and literally shakes ap art the bonds such as disulphide bridges and H -bonds b etw een segm ents o f am ino acid strings, causing the p rotein to unfold. W hen the tem p erature goes higher, the p ro tein gains m ore energy, to form new , strong er bond s w ith the neigh bo urin g m olecules. A n exam ple w ould be to boil an egg, the liquid egg w hites turns hard and opaque. C ertain chem icals cause th e Soya m ilk to b ecom e solid, and m ake the fam ous C hinese food - “T O F U ” . In som e case the proteins can refold into th eir native states i f the so lv ent is restored b ack to origin status, e.g. the tem p eratu re is low ered and certain chem icals are rem oved. In o th er
1 The denaturing condition includes: urea, guanidine hydrochloride, guanidine thiocyanate, organic solvent or elevated temperature.
Chapter 2: P r o te in a n d P ro te in S tr u c tu re
cases, th e protein s can n o t fold back, such as it is n o t po ssible to retu rn a boiled egg to its raw state, and m ake the to fu back into so ya m ilk.
P rotein m isfolding occurs as a re su lt o f p rotein co m position dam aging. F o r instance rem oval o f a num b er o f am ino acids from the p eptide chain can cause th e co llap se o f the p rotein structure and lead to arb itrary u seless spherical conform ations.
E m pirically the p rotein structure co u ld be determ ined by X -ray cry stallo graph y o r N M R spectroscopy. In silico the protein structure could be p redicted by a learning netw ork, w hich is trained previously. In th e fo llow ing paragrap h a series o f p rotein structural concepts is illustrated.
2 .4 P r o te in C o m p o s itio n a n d S tr u c tu r e s
P roteins are form ed from sequences o f am ino acids, w hich are bo nded tog eth er by covalen t bonds in a ‘head to ta il’ con figu ratio n. Som e p ro tein s contain only a single peptide chain; som e o f the m ore co m plicated p roteins co ntain m ore than one p eptide chain and form com plicated quatern ary structures. P eptide chains fold into a stable structure by inter-m olecular or intra-m o lecu lar forces, such as v an d er W aals fo rc e s2, hydrophobic fo rc e s3, H -b o n d s4etc. T he p rotein structure is dom in ated by a m ixture o f effects o f these forces. In general, p ro tein fo ld in g is an am azin g p h en o m en a th a t occurs right here on E arth.
2 In chemistry, the term van der Waals force refers to a particular class of intermolecular forces, which describes electromagnetic forces that acts between molecules or between widely separated regions of a macromolecule.
3 In chemistry, the term hydrophobic refers to the physical property of a molecule that repelled from a mass of water molecule.
4 In chemistry, the term hydrogen bond describes a special type of attractive interaction that exists between certain chemical groups of opposite polarity. The hydrogen is stronger than van der Waals forces, however much weaker than both the ionic bond and the covalent bond.
Chapter 2: P r o te in a n d P r o te in S tr u c tu re
2.4.1 A m ino A cids as B u ilding B locks o f Protein
A m ino acids are the b uilding blocks to co n struct proteins. T here are tw enty naturally occurring am ino acids, w h ich are bon ded sequentially in a “h ead -to -tail” configuration, form ing m illions o f d iffe ren t p rotein s. E ach am ino acid shares a com m o n structure o f a central tetrahedral a carb on (Cd), w h ich is co valen tly linked to both an am ino group (N H3) an d a carboxyl group (C O O H ).
H
+h 3n — c — C O O '
R
K
Figure 2-4: Structure of amino acid (showing zwitterionic structure)
A s illustrated in th e above diagram (Figure 2-4), am ino acids are differen tiated from each oth er by the ‘R ’ residue - the side chain. F or instance, alan in e has a m ethyl group attached as the ‘R ’ residue; g lycine has a single hydrogen atom attached as its ‘R ’ residue. T he 4R ’ residue defines th e characteristics o f the am ino acid, i.e. w h e th e r it is a w eak acid o r w eak base, a hy dro p h o b ic residue or h y drop hilic residue, a p o lar residue or a n on -po lar residue etc. It is b elieved th a t th e ch aracteristics o f th e ‘R ’ resid ue are critical for protein structures and functions (H ill & D eG rad o 2 0 00 ;S ch ueler-F urm an & B aker 2003). A s th e 26 letters in the E n glish language form the endless literature o f poetry, opera, m asterpieces etc, the 20 am ino acids in nature, form the book o f life.
H
- H3M— G — C O O - 1
c h3
Figure 2-5: S tructure of alanine (showing Figure 2-6: Structure of glycine (showing
zwitterionic structure) zwitterionic structure)
H
I
H 3N — C — COO"
1
Chapter 2: P ro te in a n d P r o te in S tr u c tu re
A m ino acids jo in tog eth er via p ep tid e bonds. T he am ino g roup and the carboxyl group o f each am ino acid m ay re act w ith each o th er in a ‘h ead -to -tail’ fashion by d eh yd rating one w ater m olecule, and fo rm in g a co v alen t linkage. C ertainly, at both ends o f the protein, there rem ains a free am ino group, and a carboxyl group, w hich are referred to as the N
and C term ini. The C -term inus o f p roteins, w hich includes the single term inal alpha- carboxyl group and preced in g residues, is uniquely po sitioned to serve as a recognition signature fo r a variety o f cell biolog ical p ro cesses, such as p rotein targeting , su bcellular anchoring and static and dynam ic form ation o f m acrom o lecu lar com plexes (C h un g et al.
2002). H ow ever the study o f term ini is n o t included in this thesis. T w o linked am ino acids are called dipeptides, th ree am ino acids are nam ed tripep tides, and fo u r am ino acids are called tetrapeptides. L o n g chains o f am ino acids also are called olig op eptide or p olypeptides, or p roteins. A n exam p le o f a trip eptid e is represented in the follow ing Figure 2-7: T H H 1 4- 1 i ^ c o V h
3
nr t N c c o o
-R 1 X - N R 2 r 3\
P e p t i d e B o n dFigure 2-7: A tripeptide with two peptide bonds (showing zwitterionic structure)
A s the dehydration reaction causes th e linkage o f p eptide bonds, the am ide hydrolysis reaction (the addition o f w a te r m olecular) breaks the peptide bonds. T he process o f debonding is extrem ely slow in living organism s, and is u sually cataly zed by enzym es. F u rth er reading ab o u t pep tid e bonds can be found in (P auling l9 3 l;P a u lin g l9 3 2 ;P a u lin g
l9 8 5 ;P a u lin g & S herm an l9 3 3 b ;P a u lin g & S herm an l9 3 3 a ;P a u lin g & W h eland 1933).
2.4.2 P rim ary Stru ctu re as Sequence Indicator
T he prim ary stru cture o f a p rotein is rep resented by the am ino acid sequence w ithin the polypeptide chain(s). In the prim ary structure o f a particular protein, only the identities o f am ino acids are declared. T h ere is no structural inform ation carried w ith in the prim ary
Chapter 2: P ro te in a n d P ro te in S tr u c tu re
structure o f a protein, unlike concepts such as the secondary structure, o r the tertiary structure o f a protein, m ainly focus in presentin g three dim ensional conform ational structures o f it.
In the follow ing diagram Figure 2-8 a snapshot o f the protein prim ary structure is presented:
T he term “prim ary stru ctu re” w as firstly coined by Kaj Ulrik Linderstrom-Lang in his 1951 Lane Medical Lecture: “Proteins and Enzymes'
2.4.3 Secondary Structure as Pattern o f G eneral 3D Form
The secondary structure is used to describe the captured general patterns o f three dim ensional form s o f a local segm en t in the polypeptide chain. T he form al definition o f the secondary structure relies on the patterns o f hydrogen bonds betw een backbone am ide groups. The com m only used secondary structure types o f the D ictionary o f Protein S econdary S tructure (D S S P ) are defined according to above definition (H ooft et al.
1996;K absch & S ander 1983b):
Figure 2-8: Protein prim ary structure. (G NU L icensed)
• G = 3-turn helix ( 3 1 0 helix). M inim um length 3 residues.
Chapter 2: P r o te in a n d P r o te in S tr u c tu re
• H = 4 -turn helix (« helix). M inim um length 4 residues. • I = 5-turn helix (pi helix). M inim um len gth 5 residues. • T = hydrogen bonded turn (3, 4 or 5 turn).
• E = 0 sh eet in parallel an d /o r anti-parallel sheet co nform atio n (extended strand). M in length 2 residues.
• B = residue in isolated //-bridge (single p air //-sh eet hy drog en bond form ation). • S = bend (the only non -hyd ro g en -b o n d based assignm ent).
T he rem aining bits are classified as coil, stated as “random co il” . In secondary structure prediction, there are th ree secondary structural units, w h ich nam ed *a h e lix ’, ‘// stra n d ’, and ‘tu rn ’, w idely used in the literature. A t the m om ent, helix, strand and coil are the th ree secondary structure units th a t ap plied in m ost o f p rotein secondary structure p rediction applications. A p art from the eigh t secondary structures defined in the D SSP program m e (K absch & S and er 1983a), there are several o th er secondary structure clarification m etho dologies in trodu ced in the literature, such as D E F IN E (R ichards & K u n d ro t 1988) and S T R ID E (F rishm an & A rg os 1995), w h ich are all heavily cited in the literature. T here are review s in the literature d escribing the app licatio ns uses o f these secondary structure alignm ents (F rishm an & A rgos 1997a;F rishm an & A rgos 1997b;Frishm an & A rgos 1996b).
To sim plify the problem , usually only th ree o f secondary structure units are applied in the m ethod ologies to pred ict protein seco nd ary structure: helix, strand, and coil (H, E, and C). T here are several eig h t to th ree reduction m ethods to be app lied to redu ce the com plexity:
1. H, G and I to H; E to E; th e rest to C. 2. H, G to H; E, B to E; the rest to C. 3. FI, G to H; E to E; the rest to C. 4. H to H; E, B to E; the rest to C. 5. H to H; E to E; the rest to C.
Chapter 2: P ro te in a n d P ro te in S tr u c tu re
It is obvious that the m ethod 5 increases the accuracy (C u ff & B arton 2 0 0 0 ;C u ff & B arton 1999). The PA M S applied m ethod 1 to provide a strict m easu rem en t to the prediction accuracy.
In the follow ing Figure 2-9, a three dim ensional structure o f the m yoglobin protein is presented. The coloured am ino acids segm ents present a helices, and the coils are presented in w hite. T here are no 0 strands in this protein. T his protein w as the first to have its structure solved by X -ray crystallog raph y by M ax P erutz and Sir John C ow dery K endrew in 1958 (K E N D R E W & PE R U T Z 1957). T his discovery brought them the N obel Prize in C hem istry in 1962.
Figure 2-9: T h ree dim en sion al stru ctu re o f the m yoglobin protein. (G NU L icensed)
The research into protein secondary structure prediction is w idely published in the literature. It is considered to be the first step tow ards the ultim ate goal o f protein structure determ ination (B aldi et al. 1999;Frishm an & A rgos 1997b). T his thesis presents an effective fram ew ork to process the biological sam ples, in o rd er to achieve b etter protein secondary structure prediction accuracy regardless o f the m achine learning schem es used.
Chapter 2: P r o te in a n d P ro te in S tr u c tu re
2.4.4 Super-Secondary Structure
In 1973 the discovery w as m ade th a t a few a-h elices and + -strands w ere frequently repeated w ithin structures, nam ed “su p er-seco nd ary stru ctu re” (R ao & R ossm ann 1973). It w as suggested that th ese structural m o tifs w ere associated w ith p articu lar b iological functions, w hile others w ere p art o f a big structure and fo rm ed overall biological functions. A ccording to the classificatio n (L ev itt & C hothia 1976), th ere are fo ur m ajo r groups:
• proteins con taining m ostly a-h elices; • proteins con tain in g m o stly + -sheets;
• proteins th at contain a-h elices and ^ -sh e e ts in an irreg ular sequence; • a/0 p roteins w ith alternate segm ents o f cc-helices and + -sheets.
T he sim plest m o tif is tw o helices jo in e d by a loop, w h ich is respo nsib le fo r D N A binding, and calcium binding, th ereb y reg u latin g cellular activities (P rocyshyn & R eid 1994a;P rocyshyn & R eid 1994b;R eid & P ro cysh yn 1995).
2.4.5 T ertiary Structure
T he tertiary structure o f a p rotein defines its actual shape. It co ntains the co nform ational inform ation o f all atom s w ith in it w ith o u t considerin g its relationsh ip s w ith o ther m olecules. K now ing the tertiary structure o f a p rotein is essential in the study o f understanding a p ro te in ’s biological functions. C urrently, th e d eterm ination o f tertiary protein structure is hig h ly d ep en d en t on the p articip ation o f X -ray crystallog raph y tech nique or m u lti-dim ension N M R spectroscopy. H ow ever, som etim es the screening m ethodologies are not accurate and even could n ot d eterm ine som e bits o f the p ro tein structure, e.g. X ray d iffraction gives th e atom s in po sitio n in space, but th ey have to be in a crystal. H ow ever som e bits o f the protein cannot be cry stallised such as those resp onsible for connection bits in the p ro tein o f H N F4 (alpha) o r H epatocyte N u c le ar F acto r-4a, w hich are constan tly ch an ging and thus not be seen p rop erly (A ggelidou et al. 2 0 0 6 ;D he-P agan on et al. 2002;D uda, C hi, & S hoelson 2004). T he p rediction m ethods w ill
Chapter 2: P ro te in a n d P ro te in S tr u c tu re
help in this kind o f situation. O b tain in g high prediction accuracy for the secondary structure is the first step tow ards predictin g tertiary structure.
The tertiary structure o f a protein m ay contain m ore than one peptide chain. In the follow ing Figure 2-10, an insulin sam ple w ith tw o chains is presented. C hain A contains 21 am ino acids and chain B contain s 30 am ino acids. T hese tw o chains are bonded to g eth er w ith tw o disulphide bonds to establish a reasonable stable structure for the insulin.
Figure 2-10: Stick representation of overall structure of insulin.
T he tertiary structure o f this protein is m aintained by the protein core, w hich is form ed by the hydrophobic residues oriented inside, and hydrophilic residues staying outside (E sipova & T um anyan 1972). T he disu lfide bridges, form ed betw een tw o cysteine residues, are another m ajor force in the preservation o f the tertiary structure.
Chapter 2: P r o te in a n d P ro te in S tr u c tu re
2.4.6 Q uaternary S tructure
T he quaternary structure is an even m ore co m p licated p rotein structure, form ed by polypeptide chains in space and re lian t on no n -co v alen t interactions. The p roteins th at could have quaternary structures m ust have no chains w hich are covalen tly associated. A typical exam ple is haem o glob in th a t con tain s fo ur polypeptide ch ain s held to g eth er in a specific conform ation as required fo r its fun ctio n (K ilm artin, H ew itt, & W ootton 1975). B oth tertiary structure and quaternary stru ctu re are m entioned by structural b iologists quite often in the literature. A better u n d erstanding o f tertiary structure and quatern ary structure brings new insight fo r the d iscov ery o f new drugs to attack d iseases, w hich are currently incurable, such as cancer.
2.5 Essentiality o f Protein Structure
T he research o f p rotein structure has b eco m e the centre o f p h arm aceu tical research becau se o f the p rom ising future. B etw een D ecem b er 1995 and M arch 1996, the F ood and D rug A dm inistration (F D A ) o f U nited S tates o f A m erica ap p ro v ed the first three H IV protease inhibitors - H offm an-L a R o c h e ’s Invirase™ (saqu inav ir), A b b o tt’s N o rv ir™ (ritonavir), and M erck and C o., In c .’s C rix iv an® (indinavir). T hese m ed icin es have d rastically cut the num bers o f A ID S deaths in developed countries in th e last few years, and although these H IV p ro tea se inhibitors did not becom e the m iracle cure they expected to be, they also represent a triu m p h o f dev elo p in g new drugs.
T he ultim ate goal o f curing g enetic d iseases and d eveloping in divid ual-o riented therapies requires a fast, econom ic instrum ent to determ ine th e structure and m o v em en ts o f the co rresponding faulty protein. B y m ak in g th e m alfu nction ed p ro tein w o rk , the disease m ay be cured.
2.6 Conclusions
Protein is life. T he phrase h as been ackn o w led g ed fo r o ver one h un d red years, b u t the un d erstanding o f pro tein structure is in su fficien t for industrial d em ands. H o w ev er a new w av e o f b reakthroughs in this area is b eing form ed at the m om ent, and a large n u m b er o f
Chapter 2: P r o te in a n d P r o te in S tr u c tu re
review s suggest th at the g o ld en age o f p ro tein is com ing (B ai & E n gland er 1996;B aldi et al. 1999;C hen et al. 2005 ;C h o p ra 1998;D uncan, R in gsdo rf, & S atehi-F ainaro 2006;G eisow 1992;Johnson-L eger et al. 2 006;K atzen, C hang, & K ud lick i 20 05;K o ehl & L ev itt 1999;M iddaugh 1994;M yles 2 0 0 6 ;R o senb erg & G oldblum 20 06;S h ehad i, Y ang, & O ndrechen 2002;T ram ontano 20 04 ;T su n asaw a 1994;W aterlow 1995). P rogress in u nderstanding both structural and functional characteristics o f the p ro tein is essential and m u st be achieved.
T he next chapter introduces the state o f art o f protein structure pred iction , m ore precisely, secondary structure prediction.
Chapter j . P ro te in S e c o n d a ry S tr u c tu re P re d ic tio n
3 Protein Secondary Structure Prediction
S ince L evinthal observed th at there w as insufficient tim e to search the entire conform ational space exhau stively fo r appropriate protein structure candidates, people realized that the prediction o f protein structure has to be approached in an alternative w ay. T he techniques applied in p redicting protein structures are m ulti-d isciplinarily diverse, such as evolution theory, data m ining, and m achine learning. In the past fo ur decad es the study o f protein secondary structure has received a great m any attentions in the literature. C urrently, the highest prediction accuracy is approxim ately 78% (R ost & S ander 2000).
3.1 Overview of General Protein Structure Prediction
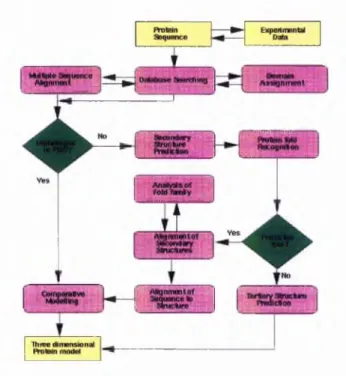
The process o f protein 3D structure prediction at the atom ic level could be defined by follow ing the follow ing flow chart in (Figure 3-1):
Figure 3-1: Protein structure prediction flowchart (by Rob Russell, EMBL)
Chapter J .P r o t e i n S e c o n d a r y S tr u c tu re P r e d ic tio n
A s described in th e ab ove flow chart, once th e protein sequence is identified from the experim ental data, it can be ap plied by m ultiple sequence align m ent (M S A ) straightforw ardly. The M S A m eth od resu lts w h eth er the input sequences have an evolutionary relation ship by w h ich they share a lineage and are d escend ed from a co m m on an cestor (B acon & A n d erso n 1986). T he M S A m etho ds are com m only used to assess sequence conservation o f p ro tein dom ains, protein secondary structure, and p rotein tertiary structure. M ore and m ore sop histicated algorithm s have been im plem ented in im proving M S A accuracy such as th e im plem entatio n o f Fuzzy H id den M arkov M odels, B ay esian segm ental m odels etc (C hu e t al. 2 00 6;C ollyd a e t al. 2 0 06 ;M o rg enstern e t al. 2006;Pei & G rishin 200 6 ;R o sh an & L iv esay 20 06;Y am ada, G otoh, & Y am ana 2006). S im ilar to th e p ro tein secondary structure prediction , v ariou s m achine learning m odels have been applied. O nce the sequence is identified to be h om o lo go us to a fam ily o f proteins w ith know n structures co m parative m odelling tech n iq u es (C enteno, P lanas- Iglesias, & O liva 2 0 0 5;C o ntreras-M o reira, F itzjohn, & B ates 20 02 ) can be used to produce a th ree dim ensional p ro tein m odel. W hile the sequence shares no o bvious hom ologous ch aracteristic w ith pro tein s w ith k now n structure, the th ree dim ensional protein m odels have to be p red icted by follo w in g an altern ative route. T he first step tow ard th at route is to p red ict the secondary structure o f th e sequence. T his p rediction provides the location o f a h elices and p sheets. O nce a secondary structure o f pro tein is pred icted both the sequence and seco nd ary structure are fed into a fold recognition program . The theo retical foundation o f th is p rocedu re is th at som e o f the p ro tein s m ay n ot share significant sequence sim ilarity, b u t th ey still share co m m o n structures (G odzik 2003 ;K oretke et al. 1999;M arch ler-B au er & B ryan t 1999;O ta 2002). T he fold recognition program s tend to d etect sim ilarities b etw een protein 3D structures, b ut require extra structural inform atio n generated by the first step. I f there are no ob vious folds recog nized, the p rotein has to go to ab initio structure p red ictio n server (B rad ley et al. 2 0 0 5 ;B y stro ff & B ak er 1 9 97;B y stroff & B aker 1998;R ohl et al. 2004), and end up h avin g a structure p redicted ov er there. I f th e re su lt is po sitiv e th a t th e protein ad op ts a p articu lar fold w ithin the database, a proced u re o f analyzing th e fold fam ily is req uired to both v alid atin g the fold results and p erform an alig n m en t o f the secondary structure elem ents to the p rotein and the found proteins th at ado p t a sim ilar fold. T he SC O P, C A T H , F SS P protein structure classification d atabase could be used in this p ro ced u re (G reene et al. 2 006;H adley & Jones 1999;H olm & S an der 1996;R eedy & B ourne 2003). T he suitable
Chapter 3 /P r o te in S e c o n d a ry S tr u c tu re P r e d ic tio n
fold via fold recognition can be fed into the hom ology m odelling tech niqu es to be used to deduce the tertiary structure o f the giv en protein. The ho m ology m odellin g techniques includes W H A T IF, M O D E L L E R , etc (F iser & Sali 2003 ;V rien d 1990).
In order to p re d ic t the tertiary structure from sequence alone, th e p red ictio n o f secondary structure is the initial step. W ithout a p red ictio n o f secondary stru ctu re the pu re ab initio
p rediction o f tertiary structure from sequence alone is v ery com pu tation ally expensive. In th e follow ing parag rap h a literatu re rev iew o f protein secondary structure pred ictio n is presented.
3.2 Historical View o f Protein Secondary Structure Prediction
H istorically speaking the first attem p t to determ ine the secondary structure o f a p rotein from its sequence w as p erform ed in 1957, try in g to co rrelate the proline w ith the form ation o f a helix (S Z E N T -G Y O R G Y I & C O H E N 1957), sh ortly after P au lin g and C orey suggested the existence o f the a helix and 0 sheet (P A U L IN G & C orey 1951a;P A U L IN G & C orey 1951b;P A U L IN G &