S O F T W A R E R E V I E W
Open Access
MEGADOCK 3.0: a high-performance
protein-protein interaction prediction software
using hybrid parallel computing for petascale
supercomputing environments
Yuri Matsuzaki
1,3*, Nobuyuki Uchikoga
2,3, Masahito Ohue
1,4,5, Takehiro Shimoda
1,4, Toshiyuki Sato
6,
Takashi Ishida
1and Yutaka Akiyama
1,3,4Abstract
Background:Protein-protein interaction (PPI) plays a core role in cellular functions. Massively parallel
supercomputing systems have been actively developed over the past few years, which enable large-scale biological problems to be solved, such as PPI network prediction based on tertiary structures.
Results:We have developed a high throughput and ultra-fast PPI prediction system based on rigid docking,
“MEGADOCK”, by employing a hybrid parallelization (MPI/OpenMP) technique assuming usages on massively
parallel supercomputing systems. MEGADOCK displays significantly faster processing speed in the rigid-body docking process that leads to full utilization of protein tertiary structural data for large-scale and network-level problems in systems biology. Moreover, the system was scalable as shown by measurements carried out on two supercomputing environments. We then conducted prediction of biological PPI networks using the post-docking analysis.
Conclusions:We present a new protein-protein docking engine aimed at exhaustive docking of mega-order numbers of protein pairs. The system was shown to be scalable by running on thousands of nodes. The software package is available at: http://www.bi.cs.titech.ac.jp/megadock/k/.
Keywords:Parallel computing, Protein docking, Protein-protein interaction network, Protein-protein interaction prediction
Background
Living cells are maintained by a multitude of molecular interactions. The regulatory interactions among the thousands of proteins (Protein-Protein Interaction, PPI) in a human cell are currently being elucidated. A detailed knowledge of such interactions will be crucial for understanding the mechanisms that underlie diseases and for the development of a new generation of drugs.
The next challenge is to perform interactome level large-scale analysis by fully utilizing protein tertiary structures. In order to address this problem we proposed a large-scale PPI prediction method based on exhaustive protein docking and post-docking analysis [1-3]. Using this system, we input protein tertiary structure data to acquire predictions of possible interacting pairs.
For example, when reconstructing the human apoptosis pathway [4], 158×158 potential combinations of structures were considered. We have already applied our system to 44×44 (subset of Protein-Protein Docking Benchmark 2.0 [5]) and 89×89 (structures of bacterial chemotaxis proteins) scale analyses [1,6]. In real biology problems, such as searching for the drug induced pathway of EGFR (Epidermal Growth Factor Receptor) signaling, about * Correspondence:[email protected]
1Graduate School of Information Science and Engineering, Tokyo Institute of
Technology, 2-12-1 Ookayama, Meguro-ku, Tokyo 152-8550, Japan 3Grand Challenge Applications Project for Life Sciences, Next-Generation
Integrated Simulation of Living Matter, Computational Science Research Program, Riken, 2-1 Hirosawa, Wako, Saitama 351-0198, Japan Full list of author information is available at the end of the article
200 proteins need to be examined. In our preliminary survey on the EGFR pathway and related proteins data, we identified about 2000 structures corresponding to these proteins. Therefore, the PPI network prediction system needs to handle about 2000×2000 combinations of protein structures.
To solve such large-scale problems, a highly efficient computing system is necessary. High performance com-puters are currently being developed and built [7]. Some top ranked supercomputers have shown a peak per-formance of 27 petaflops (Titan, Oak Ridge National
Laboratory, USA) and 11 petaflops (K computer, RIKEN, Advanced Institute of Computer Science (AICS), Japan) in November 2012.
We have implemented a protein docking system
“MEGADOCK”suitable for running on supercomputers by using hybrid parallelization (MPI, Messeage Passing Inter-face/OpenMP, Open Multi-Processing), where a number of docking processes are distributed among the nodes by MPI with each docking process also calculated in parallel by threads by OpenMP within one node. Data parallelization showed almost linear scaling up to 24,576 nodes on K
computer (RIKEN AICS, Japan). In addition, we also designed MEGADOCK using a simple score model to re-duce the number of calculations required for protein docking.
We expect the proposed method can be a useful tool in bioinformatics and systems biology area as a basic tool, assuming we can utilize ~10,000 CPUs.
Implementation
We adopted two strategies to make MEGADOCK suitable for large-scale docking simulations performed on super-computers. First we devised a novel score function with real Pairwise Shape Complementarity, rPSC that enables us to calculate two aspects of interactions using one convolution function with Fast Fourier Transform (FFT) calculations. This setup markedly reduces the time required for each docking calculation per protein pair. Second, we implemented MEGADOCK by hybrid paralle-lization (MPI/OpenMP) in order to conduct large numbers of docking jobs for PPI network predictions.
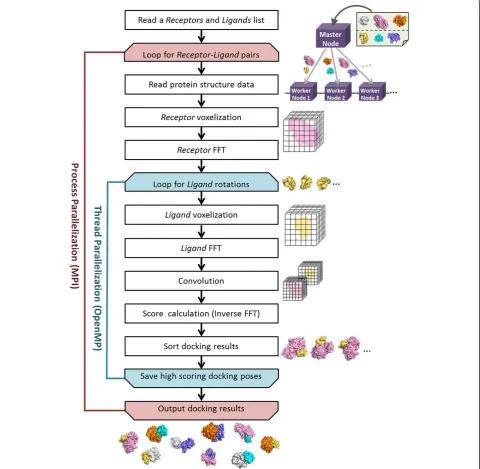
The overall procedure of MEGADOCK is shown in Figure 1. On the cluster computers, a master node gets a list of protein structures and distributes the docking jobs to available nodes. Upon docking of two proteins (here we call them “receptor” and “ligand”, apart from biological definition, to indicate two docked proteins), a
“ligand”is rotated to various orientations and translated in the space around the“receptor”, which is fixed during docking calculation, to search for the best scoring positions. These processes are parallelized by threads.
Docking score function
MEGADOCK includes a rigid-body docking system that searches relevant interacting poses between the target protein tertiary structures. The rigid-body docking process is mainly based on shape complementarity of the target proteins without considering conformational changes upon formation of the complex structures.
Docking scores are defined by an equation consisting of the shape complementarity term G and the electrostatic interaction term E. The target protein pairs R (receptor) and L (ligand) are first allocated on the 3-D voxel spaceV, 1.2Å × 1.2Å × 1.2Å. Then the voxel values are assigned to each voxel (l,m,n)∈Vaccording to the location in a protein, open space and core. We use our original scor-ing function called“real Pairwise Shape Complementar-ity” (rPSC) for the shape complementarity term G as follows [2]:
GLðl;m;nÞ ¼ 01 ðopen spaceðcore of LÞÞ
Here rvdw represents the van der Waals radius of an atom, and (α,β,γ) is a vector of the ligand translation.
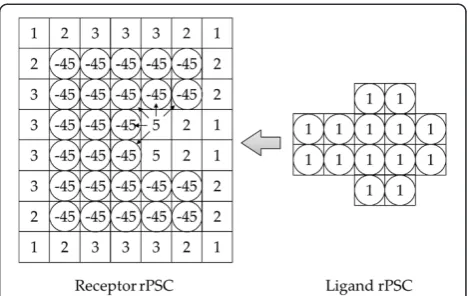
The advantage of rPSC is that it is a real number repre-sentation (Figure 2), thus we can put a physicochemical parameter into the imaginary part of the voxel score, which are complex numbers. Hence, it is possible to calculate docking scores by assigning only one complex number for each voxel.
For physicochemical parameters, we use the electrostatic interaction of each amino acid, ER(l,m,n) and EL(l,m,n). These values are calculated using the interaction energy in each voxel,q (l,m,n) using CHARMM19 [8] for the electrostatic charge of each atom.
Considering these terms, the overall docking scoreSis calculated as follows:
R l;ð m;nÞ ¼GRðl;m;nÞ þiERðl;m;nÞ
L l;ð m;nÞ ¼GLðl;m;nÞ þiwELðl;m;nÞ
Sðα;β;γÞ ¼R X
N
l¼1
XN
m¼1
XN
n¼1
R lð;m;nÞL lð þα;mþβ;nþγÞ
" #
For direct execution of a simple convolution sum, O (N6) calculations are required. The calculation order using fast Fourier transform (FFT) algorithms, both for a discrete Fourier transform (DFT) and inverse discrete
Figure 2Real pair-wise shape complementarity score model.
For each potential docking process, the sums of the overlapped voxel values are assigned as the docking score.
Fourier transform (IFT), is reduced to O(N3logN) [9]. The scoreSfor FFT is:
Sðα;β;γÞ ¼R½IFT DFT½ ½R l;ð m;nÞDFT½L l;ð m;nÞ
By reducing the number of required DFT/IFT operations with rPSC, MEGADOCK in theory should perform the docking process faster than the well-known protein-protein docking tool, ZDOCK [10,11], which uses eight complex numbers per calculation.
Hybrid parallelization
The overall procedure of MEGADOCK is shown in Figure 1. Initially, a master node distributes docking jobs to available nodes after obtaining a list of protein pairs. We parallelized the calculation of each docking processes using MPI library (Figure 1, red colored loop). After one loop of this MPI, we obtain high scoring poses among all the rotation and translation patterns of assigned protein pairs.
Each docking process in each node is parallelized to threads by OpenMP (Figure 1, blue colored loop). Upon docking, the coordinates of the ligand are repeatedly rotated and translated to search for a better complex form with the receptor. The calculations of FFT and inverse FFT for each rotation angle are performed independently. Thus, using OpenMP loop we calculate high scoring poses for various rotation angles in parallel.
The implementation is designed to run efficiently on K computer which has 88,128 nodes with 8 cores per node (i.e., total of 705,024 cores). Each node is equipped with 16 GB of memory. Flat MPI is often used for parallel applications. However, using flat MPI on numerous core systems like K computer may result in a large overhead due to handling data communication of ~700,000 cores. Thus, hybrid parallelization is efficient on such high performance computing systems.
Reducing usage of memory space is important with systems that have many cores per node and relatively small memory size. In flat MPI, the docking job of each protein pair is assigned to each core. Thus, each core requires memory space for input/output data. If a node has n cores, the memory space in the node should be large enough to keep data fornpairs of pro-teins (in the case of K computer, n = 8). In contrast, by implementing hybrid parallelization, we assign one protein pair to each node and then distribute the cal-culations of ligand rotation by thread parallelization. As such, each node will keep data of one pair of proteins on the memory and threads will share the input/output data on the memory. The memory size needed for docking is dependent on protein size. This implementation is feasible when considering calculations of large pro-teins. Thus, we implemented MEGADOCK by hybrid parallelization.
Results and discussion
Dataset
We used a general benchmark dataset for protein docking (Protein-Protein Docking Benchmark 4.0, [12]) to evaluate the accuracy of predicted docking poses generated by MEGADOCK. This benchmark set comprises 176 known complexes and included both a“bound”and“unbound”set. The“bound”set is composed of protein structures prepared by separating individual proteins from the crystal structures of 176 protein complexes. The“unbound”data means that each protein structure is taken from the isolated form of crystals rather than complex form. The “unbound” dataset includes protein structural data corresponding to the same set of proteins in the “bound” dataset. Structural differences in bound and unbound form in RMSD are shown in the reference [12].
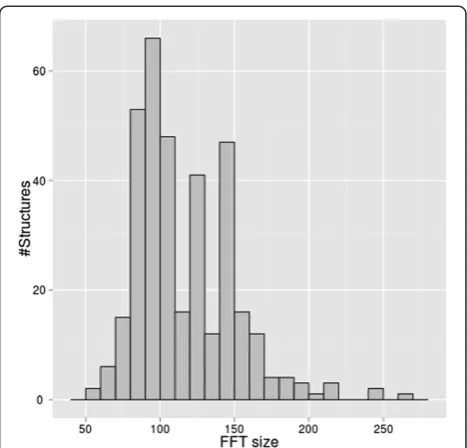
We also evaluated elapsed time for docking calculations over this dataset (calculation of 176 protein complex predictions). Distribution of size of FFT for protein in
Figure 3FFT size of various proteins in protein-protein docking benchmark 4.0 (176 Protein complexes, 352 structures).
Table 1 Total calculation time on one node and one thread to compute 176 dockings of protein-protein docking benchmark 4.0 [12]
Name Running time [hr] Speedup from ZDOCK 3.0
MEGADOCK 3.0 41.7 8.77
ZDOCK 2.3 157.3 2.32
ZDOCK 3.0 365.6 1.0
this dataset is shown in Figure 3. Time consumed for FFT is expected to be longer with larger size of FFT.
For measuring thread parallel scalability, we conducted dockings of a protein complex from PDB, 1ACB (chain E and I). The size of FFT is 108 in this case. Parallel scalability over nodes using MPI was measured by con-ducting exhaustive docking of 220 different proteins (220×220 dockings), with an FFT size of 140.
Test environment
Parallel scalability of MEGADOCK was measured on two supercomputing environments, TSUBAME (Tokyo Institute of Technology Global Scientific Information and Computing Center (GSIC), Japan) and K computer (RIKEN AICS, Japan). The most abundant node type of TSUBAME had an Intel Xeon 5670, 2.93 GHz processor, 12 cores. Each node is capable of up to 24 threads of putation by using the hyper threading technique. K com-puter has Fujitsu SPARC64 VIIIfx CPUs, 2 GHz, 8 cores.
Calculation speedup
Table 1 shows the total calculation time needed for docking 176 protein complexes in Protein-Protein Docking Bench-mark 4.0 [12]. MEGADOCK calculation speed was measured from the calculation time using one node and one thread to show the baseline. The measurement was conducted on a node of the TSUBAME supercomputing
system (Tokyo Institute of Technology, Japan), equipped with an Intel Xeon 5670, 2.93 GHz processor. In Table 1, we list the other two FFT-based docking software packages, ZDOCK 3.0 and ZDOCK 2.3. Both versions of ZDOCK use the same FFT library [13] as MEGADOCK. Ligand FFT, inverse FFT consists large part (on average 85.1% for 176 dockings) of the calculation time as shown in Table 2. As all three software listed in Table 1 uses the same FFT library (FFTW), Table 1 shows the obtained speedup by adopting the simplified score function with rPSC compared to ZDOCK 3.0.
The dataset includes proteins of various size (Figure 3). The time required for each docking calculation is de-pendent on protein size. For example, a protein that re-quires size 120 FFT calculations (1E96) gave a calculation time of about 547 seconds. Smaller sized protein pairs, such as size 80 FFT (1GCQ) were calculated in about 155 seconds. This variation in calculation time reflects the difference of FFT calculation (size 120×120×120 and 80×80×80). The smaller protein pair (size 80 FFT) takes about 0.28 times the elapsed time compared to the larger protein pair (size 120 FFT). This ratio of elapsed time is reasonable. In theory FFT takes the order ofO(N3logN) for calculation. Therefore calculations involving a size of 80 FFT should take ~0.27 times ((803log 80)/(1203log 120)) the elapsed time of a corresponding calculation involving a size of 120 FFT, which is almost the same scale as the calculation time we measured on TSUBAME.
Parallel scalability
Figure 4 shows the thread parallel scalability of MEGA-DOCK by parallelizing ligand rotation and FFT calculation. The calculation time is shown as an average of 10 individ-ual docking events with an FFT size of 108 (1ACB chain E and I) from the benchmark data. We observed a 7.33-fold speedup when using the maximum number of threads, 8 threads, on K computer compared to a single thread calculation. We observed a 9.17-fold speedup for 12 threads of calculation and a 10.42-fold speedup for 24 threads of calculation compared to a single thread calculation. Note Table 2 Ratio of time spent for each process in the total
docking time (Average of 176 dockings of protein-protein docking benchmark 4.0 [12], calculated with single thread setting)
Calculation Ratio of time spent for
the process [%] (mean±sd)
Receptor voxelization and FFT 1.19±0.62
Ligand rotation and voxelization 6.41±3.13
Ligand FFT 40.38±2.79
Inverse FFT 45.99±2.25
Sort docking results 6.02±1.46
that in TSUBAME system we measured time with hyper threading activated, so number of threads more than 12 includes slight speedup including this effect.
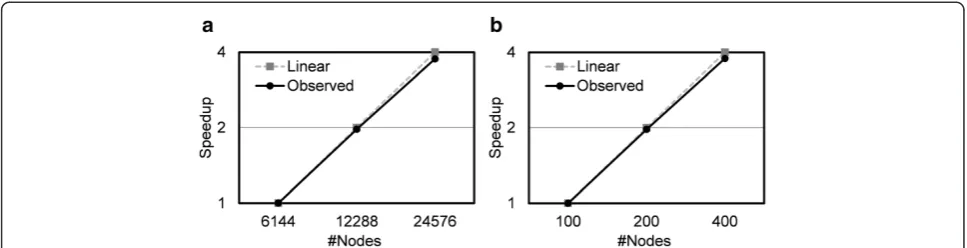
Figure 5 shows a process level parallel scalability of MEGADOCK. On K computer, where a maximum of 24,576 nodes can be used simultaneously, we measured the time needed to calculate exhaustive dockings of 220 proteins (220×220=48,000 dockings), calculated with a size of FFT 140. Calculation time using 24,576 nodes was about 3.76-fold faster than the time needed to solve the same problem on 6,144 (1/4 of 24,576 nodes) nodes. On TSUBAME, we measured the time needed to calculate exhaustive dockings of 44 proteins (44×44=1,936 dockings) using up to 400 nodes at a time. Calculation time using 400 nodes was about 3.78-fold faster than that on 100 nodes. MEGADOCK achieved almost linear scalability on both supercomputing environments.
We used a dataset of similar sized proteins (FFT size 140) for the scalability test. It is an unrealistic scenario when calculations conducted by each node are almost equal. Thus, the only possible overhead by parallelization is the job distribution and checking by the controller nodes. For real problems, which include simulating dockings of proteins with a variety of sizes, a more intelligent controller is needed to efficiently distribute docking tasks according to the protein size.
Another possible improvement to make calculation faster could be on the FFT calculation. A profiler output
showed that about 86.37% of the elapsed time was used by FFT and inverse FFT calculations. Users can switch the FFT engine to similar libraries by making small changes to the MEGADOCK source code. We have tried using FFTE [14], FFTW [13] and FFT function (dvcfm1) in CSSL2 (Fujitsu Ltd., Tokyo, Japan). All three implementations yielded equivalent docking outputs. The speed of calcula-tion differs depending on the size of the proteins. For example, CSSL2 was slightly faster (data not shown) than FFTW when applied to FFT size of 128 or other base 2 FFT calculations. By contrast, FFTW outperformed CSSL2 with docking simulations involving other sizes of protein. Thus, FFTW may be the function of choice for applications where the dataset includes proteins of various sizes.
Application to protein-protein interaction prediction Figure 6 shows the accuracy of protein docking by means of “Success rate”. Success rate of the top k high scoring docking predictions represents the percentage of protein pairs that have “correct” complex poses in the top k docking predictions. Here we define a prediction as being
“correct”if the predicted complex has a root mean square deviation of less than 5 Å for the coordinates of the ligand (L-RMSD, Root Mean Square Deviation of the coordinates of the Ligand) to the crystal structure. L-RMSD is a meas-ure between corresponding ligand structmeas-ures when the
Figure 5Scalability of parallelization among nodes by MPI on (a) K computer (6144 to 24576 nodes), 220×220 dockings of FFT size = 140 protein pairs; (b) TSUBAME (100 to 400 nodes), 44x44 dockings of FFT size = 140 protein pairs.
receptor part was superimposed between two given protein complexes.
Using the decoys obtained by exhaustive docking, we conducted prediction of interacting protein pairs. The PPI network prediction method is detailed in our previous publications [1,6]. We predicted relevant interacting proteins from 44×44=1,936 combinations by docking and post-docking analysis. We obtained an F-measure value of 0.42 for this dataset. The F-measure is the har-monic mean of precision (0 to 1, where 1 means all the predicted PPIs are confirmed as correct) and recall (0 to 1, where 1 means the prediction covered all the known correct interactions).
We also evaluated our PPI prediction system by applying it to the data from the bacterial chemotaxis pathway, which is one of the typical problems in systems biology. For this dataset we obtained an F-measure value of 0.52.
Conclusions
In this study, we implemented a high-throughput and ultra-fast PPI network prediction system“MEGADOCK” suitable for massively paralleled large-scale analysis of millions of protein combinations. The docking engine of MEGADOCK was implemented by parallelization techniques and shown to be scalable on massively parallel computing environments. MEGADOCK is ideally suited to a large-scale computing system.
Using the MEGADOCK framework, it is simple to improve score functions and test it on a large dataset. For example, incorporating desolvation effects were considered in a previous report [15] and will be included in future releases. MEGADOCK is an open-source software pack-age, making it easy for the user to apply different score functions. A further advantage is that users can change many parameters, such as the weight of each term of score functions.
Protein docking based PPI network prediction has various applications. For example, Acuner Ozbabacan et al.analyzed the human apoptosis pathway using the docking based system PRISM [16]. This problem required calculation of 158×158 combinations of structures. Further useful applications may include for example the EGFR signaling pathway (where approximately 2000×2000 com-binations of tertiary structures need to be examined), which is implicated in the onset of lung cancer and various other serious diseases.
Availability and requirements
Project name: MEGADOCK
Project home page:http://www.bi.cs.titech.ac.jp/ megadock/k/
Operating system(s): Linux Programming language: C++
Other requirements: FFTW, MPI, OpenMP License: GPLv3
Any restrictions to use by non-academics: No (licence holds)
Abbreviations
DFT:Discrete Fourier transform; EGFR: Epidermal growth factor receptor; FFT: Fast Fourier transform; IFT: Inverse discrete Fourier transform; L-RMSD: Root mean square deviation for the coordinates of the ligand; MPI: Message passing interface; OpenMP: Open multi-processing; PDB: Protein data bank; PPI: Protein-protein interaction; rPSC: Real pairwise shape complementarity.
Competing interests
The authors declare that they have no competing interests.
Authors’contributions
YM, NU and MO carried out the docking experiments and measurements of parallelization effects on K computer and TSUBAME. YM carried out measurements by using different FFT engines and packaging of the software. YM, NU and MO drafted the manuscript. MO, TShimoda, TI and TSato implemented the MEGADOCK software. YA conceived of the study, and participated in its design and coordination. All authors have read and approved the final manuscript.
Acknowledgements
Software building, application and measurements of parallelization efficiency was performed by using the K computer at RIKEN Advanced Institute for Computational Science (AICS), RIKEN Integrated Cluster of Clusters (RICC) facility and TSUBAME 2.0 at Global Scientific Information and Computing Center (GSIC) of Tokyo Institute of Technology. We are very grateful for helpful discussions with Aki Hasegawa, Yousuke Ohno and Gen Masumoto in RIKEN Advanced Institute for Computational Science. The technical assistance of Hikaru Inoue and Tomoyuki Noda in Fujitsu Co. Ltd. on using K computer is greatly acknowledged. This work was supported by the National Project“Next-generation Integrated Living Matter Simulation”of the Ministry of Education, Culture, Sports, Science, and Technology (MEXT) and HPCI System Research project (Project ID: hp120131).
Author details
1
Graduate School of Information Science and Engineering, Tokyo Institute of Technology, 2-12-1 Ookayama, Meguro-ku, Tokyo 152-8550, Japan. 2Department of Physics, Chuo University, 1-13-27 Kasuga, Bunkyo-ku, Tokyo
112-8551, Japan.3Grand Challenge Applications Project for Life Sciences, Next-Generation Integrated Simulation of Living Matter, Computational Science Research Program, Riken, 2-1 Hirosawa, Wako, Saitama 351-0198, Japan.4Education Academy of Computational Life Sciences, Tokyo Institute of Technology, 2-12-1 Ookayama, Meguro-ku, Tokyo 152-8550, Japan. 5
Research Fellow of the Japan Society for the Promotion of Science, 8 Ichibancho, Chiyoda-ku, Tokyo 102-8472, Japan.6Mizuho Information & Research Institute, Inc., 2–3 Kanda-Nishikicho, Chiyoda-ku, Tokyo 101-8443, Japan.
Received: 28 February 2013 Accepted: 7 August 2013 Published: 3 September 2013
References
1. Matsuzaki Y, Matsuzaki Y, Sato T, Akiyama Y:In silicoscreening of protein-protein interactions with all-to-all rigid docking and clustering: an application to pathway analysis.J Bioinform Comput Biol2009,7:991–1012. 2. Ohue M, Matsuzaki Y, Akiyama Y:Docking-calculation-based method for
predicting protein-RNA interactions.Genome Inform2011,25:25–39. 3. Uchikoga N, Hirokawa T:Analysis of protein-protein docking decoys using
interaction fingerprints: application to the reconstruction of CaM-ligand complexes.BMC Bioinforma2010,11:236.
4. Acuner Ozbabacan SE, Keskin O, Nussinov R, Gursoy A:Enriching the human apoptosis pathway by predicting the structures of protein-protein complexes.J Struct Biol2012,179:338–346.
6. Ohue M, Matsuzaki Y, Uchikoga N, Ishida T, Akiyama Y:MEGADOCK: an all-to-all protein-protein interaction prediction system using tertiary structure data.Protein Pept Lett. in press.
7. Top 500 supercomputer sites.http://www.top500.org.
8. Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M:
CHARMM: a program for macromolecular energy, minimization, and dynamics calculations.J Comput Chem1983,4:187–217.
9. Katchalski-Katzir E, Shariv I, Eisenstein M, Friesem AA, Aflalo C, Vakser IA:
Molecular surface recognition: determination of geometric fit between proteins and their ligands by correlation techniques.Proc Natl Acad Sci USA1992,89:2195–2199.
10. Chen R, Weng Z:Docking unbound proteins using shape complementarity, desolvation, and electrostatics.Proteins2002,47:281–294.
11. Chen R, Li L, Weng Z:ZDOCK: an initial-stage protein-docking algorithm. Proteins2003,52:80–87.
12. Hwang H, Vreven T, Janin J, Weng Z:Protein-protein docking benchmark version 4.0.Proteins2010,78:3111–3114.
13. Frigo M, Johnson SG:The design and implementation of FFTW3.Proc IEEE 2005,93:216–231.
14. FFTE: a fast Fourier transform package.http://www.ffte.jp.
15. Ohue M, Matsuzaki Y, Ishida T, Akiyama Y:Improvement of the protein-protein docking prediction by introducing a simple hydrophobic interaction model: an application to interaction pathway analysis. Lect Notes Comput Sci2012,7632:178–187.
16. Tuncbag N, Gursoy A, Nussinov R, Keskin O:Predicting protein-protein interactions on a proteome scale by matching evolutionary and structural similarities at interfaces using PRISM.Nat Protoc2011,
6:1341–1354.
doi:10.1186/1751-0473-8-18
Cite this article as:Matsuzakiet al.:MEGADOCK 3.0: a high-performance protein-protein interaction prediction software using hybrid parallel computing for petascale supercomputing environments.Source Code for Biology and Medicine20138:18.
Submit your next manuscript to BioMed Central and take full advantage of:
• Convenient online submission
• Thorough peer review
• No space constraints or color figure charges
• Immediate publication on acceptance
• Inclusion in PubMed, CAS, Scopus and Google Scholar
• Research which is freely available for redistribution
![Table 1 shows the total calculation time needed for docking176 protein complexes in Protein-Protein Docking Bench-mark 4.0 [12]](https://thumb-us.123doks.com/thumbv2/123dok_us/9161455.1912470/5.595.57.540.586.696/table-calculation-docking-protein-complexes-protein-protein-docking.webp)