organic papers
o4200
Swamy and Ravikumar C8H8N2S doi:10.1107/S1600536805037566 Acta Cryst.(2005). E61, o4200–o4202 Acta Crystallographica Section E
Structure Reports Online
ISSN 1600-5368
2-(Methylsulfanyl)-1
H
-benzimidazole
G. Y. S. K. Swamy* and K. Ravikumar
Laboratory of X-ray Crystallography, Indian Institute of Chemical Technology, Hyderabad 500 007, India
Correspondence e-mail: swamy@iictnet.org
Key indicators
Single-crystal X-ray study
T= 273 K
Mean(C–C) = 0.002 A˚
Rfactor = 0.033
wRfactor = 0.090
Data-to-parameter ratio = 13.4
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2005 International Union of Crystallography Printed in Great Britain – all rights reserved
The benzimidazole ring system in the title molecule, C8H8N2S,
is planar The imidazole ring does not exhibit a delocalized
aromatic bond system. Molecules are linked by N—H N
hydrogen bonds along the c axis. The crystal structure is
further stabilized by C—H interactions.
Comment
2-Mercaptobenzimidazole (MBI), a widely used antioxidant for rubbers and plastics, has a potent thyrotoxic effect in rats
(Kawasaki et al., 1998). Human exposure to MBI occurs
through the use of rubber products processed with this anti-oxidant for vulcanization (Airaudoet al., 1990). MBI is rather stable and might act as an environmental endocrine disrupter. Methylated derivatives of MBI for industrial supply are much less toxic (Paynteret al., 1988). Further, they are known to be efficient corrosion inhibitors for metals and alloys in various aggressive media (Donnellyet al., 1978). Keeping in view the importance of such compounds, we report here the crystal structure of the title compound, (I).
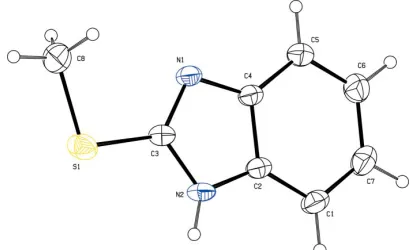
The fused ring system in (I) is planar with a maximum deviation of 0.011 (1) A˚ for the atom C3; atoms S1 and C8 are also coplanar with it. The geometrical parameters of the imidazole ring system are consistent with those in the litera-ture (Ravishankaret al., 2005). In the imidazole ring, there is significant shortening of the N1—C3 bond and lengthening of the N1—C4 bond (Table 1) compared with the values for
[image:1.610.228.433.596.721.2]Received 21 October 2005 Accepted 15 November 2005 Online 23 November 2005
Figure 1
2-mercaptobenzimidazole (Formet al., 1976); this indicates a failure to form the delocalized aromatic system which might be expected for this molecule. The C3—S1 and C8—S1 bond
lengths [1.7374 (16) and 1.783 (2) A˚ ] possess 36% and 19%
SCF- bond character, respectively (Trinajstic, 1968). The
methyl substituent at atom S1 has an impact on the bond lengths and angles of the five-membered heterocycle. This can be seen clearly by comparing S1—C3—N2 and S1—C3—N1 (Table 1) with the corresponding angles in structures with no substitution at the S atom (Kitanoet al., 1991; Ravikumar et al., 1995).
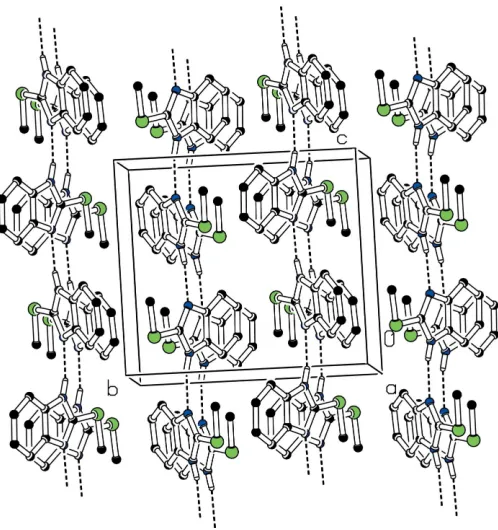
The molecules are linked by N—H N hydrogen bonds,
forming chains along thecaxis (Fig. 2). The structure is further
stabilized by a C—H interaction involving the methyl
H8Aatom and the nine-membered benzimidazole ring system
(Table 2).
Experimental
The title compound was obtained from Sigma–Aldrich and recrys-tallized from methanol.
Crystal data
C8H8N2S Mr= 164.22
Monoclinic,P21=c a= 6.9279 (4) A˚
b= 11.4613 (7) A˚
c= 10.1980 (7) A˚ = 99.052 (1) V= 799.66 (9) A˚3 Z= 4
Dx= 1.364 Mg m
3
MoKradiation Cell parameters from 2320
reflections = 2.7–27.8
= 0.33 mm1 T= 273 (2) K Block, colourless 0.220.180.16 mm
Data collection
Bruker Smart APEX CCD area-detector diffractometer !scans
Absorption correction: none 7479 measured reflections 1405 independent reflections
1298 reflections withI> 2(I)
Rint= 0.021
max= 25.0 h=8!8
k=13!13
l=12!12
Refinement
Refinement onF2 R[F2> 2(F2)] = 0.033 wR(F2) = 0.090 S= 1.03 1405 reflections 105 parameters
H-atom parameters constrained
w= 1/[2
(Fo2) + (0.0499P)2 + 0.2519P]
whereP= (Fo2+ 2Fc2)/3
(/)max< 0.001 max= 0.22 e A˚
3 min=0.20 e A˚ 3
Table 1
Selected geometric parameters (A˚ ,).
C2—N2 1.378 (2)
C3—N1 1.3160 (19)
C3—N2 1.3584 (19)
C4—N1 1.3931 (19)
N1—C3—S1 126.90 (11) N2—C3—S1 119.56 (11)
C1—C2—C4—N1 178.88 (14) S1—C3—N2—C2 176.79 (10)
N2—C3—S1—C8 176.47 (13)
Table 2
Hydrogen-bond geometry (A˚ ,).
D—H A D—H H A D A D—H A
N2—H2N N1i
0.84 (2) 2.07 (2) 2.8769 (17) 160 (2) C8—H8A Cg1ii
0.96 2.71 3.58 150
Symmetry codes: (i)x;yþ3 2;z
1
2; (ii)xþ2;yþ 1 2;zþ
1
2.Cg1 is the centroid of the
benzimidazole ring system.
The H atom attached to the N atom was located in a difference density map and refined freely. All other H atoms were placed in geometrically idealized positions and allowed to ride with C—H distances in the range 0.93–0.97 A˚ , and withUiso(H)=1.2–1.5Ueq(C).. Data collection:SMART(Bruker, 2001); cell refinement:SAINT
(Bruker, 2001); data reduction:SAINT; program(s) used to solve structure:SHELXS97(Sheldrick, 1997); program(s) used to refine structure: SHELXL97 (Sheldrick, 1997); molecular graphics:
ORTEP3(Farrugia, 1997) andPLATON(Spek, 2003); software used to prepare material for publication: SHELXL97 and PARST
(Nardelli, 1995).
The authors thank Dr J. S. Yadav, Director, IICT, Hyder-abad, for his kind encouragement.
References
Airaudo, C. B., Gayte-Sorbier, A., Momburg, R. & Laurent, P. (1990). J. Biomater. Sci. Polym. Ed.1, 231–241.
Bruker (2001).SAINT(Version 6.28a) andSMART(Version 5.625). Bruker AXS Inc., Madison, Wisconsin, USA.
Donnelly, B., Downie, T. C., Grzeskowiak, R., Hamburg, H. R. & Short, D. (1978).Corros. Sci.18, 109–116.
Farrugia, L. J. (1997).J. Appl. Cryst.30, 565.
Form, G. R., Raper, E. S. & Downie, T. C. (1976).Acta Cryst.B32, 345–348.
organic papers
Acta Cryst.(2005). E61, o4200–o4202 Swamy and Ravikumar C
[image:2.610.47.296.69.334.2]8H8N2S
o4201
Figure 2Kawasaki, Y., Umemura, T., Saito, M., Momma, J., Matsushima, Y., Sekiguchi, H., Matsumoto, M., Sakemi, K., Isama, K., Inoue, T., Kurokawa, Y. & Tsuda, M. (1998).J. Toxicol. Sci.23, 53–68.
Kitano, Y, Ishitani, A., Sato, H., Imamura, S. & Ashida, T. (1991).Acta Cryst.
C47, 1269–1271.
Nardelli, M. (1995).J. Appl. Cryst.28, 659.
Paynter, O. E., Burin, G. J., Jaeger, R. B. & Gregorio, C. A. (1988).Regul. Toxicol. Pharmacol.8, 102–119.
Ravikumar, K., Chandra Mohan, K., Bidyasagar, M. & Swamy, G. Y. S. K. (1995).J. Chem. Crystallogr.25, 325–329.
Ravishankar, T., Chinnakali, K., Arumugam, N., Srinivasan, P. C., Usman, A. & Fun, H.-K. (2005).Acta Cryst.E61, o1184–o1186.
Sheldrick, G. M. (1997). SHELXS97 and SHELXL97. University of Gottingen, Germany.
Spek, A. L. (2003).J. Appl. Cryst.36, 7–13. Trinajstic, N. (1968).Tetrahedron Lett.9, 1529–1532.
organic papers
o4202
Swamy and Ravikumar Csupporting information
sup-1 Acta Cryst. (2005). E61, o4200–o4202
supporting information
Acta Cryst. (2005). E61, o4200–o4202 [https://doi.org/10.1107/S1600536805037566]
2-(Methylsulfanyl)-1
H
-benzimidazole
G. Y. S. K. Swamy and K. Ravikumar
2-(Methylsulfanyl)-1H-benzimidazole
Crystal data
C8H8N2S
Mr = 164.22
Monoclinic, P21/c
Hall symbol: -P 2ybc
a = 6.9279 (4) Å
b = 11.4613 (7) Å
c = 10.1980 (7) Å
β = 99.052 (1)°
V = 799.66 (9) Å3
Z = 4
F(000) = 344
Dx = 1.364 Mg m−3
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 2320 reflections
θ = 2.7–27.8°
µ = 0.33 mm−1
T = 273 K Needle, colourless 0.22 × 0.18 × 0.16 mm
Data collection
Bruker Smart APEX CCD area-detector diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
ω scans
7479 measured reflections 1405 independent reflections
1298 reflections with I > 2σ(I)
Rint = 0.021
θmax = 25.0°, θmin = 2.7°
h = −8→8
k = −13→13
l = −12→12
Refinement
Refinement on F2
Least-squares matrix: full
R[F2 > 2σ(F2)] = 0.033
wR(F2) = 0.090
S = 1.03 1405 reflections 105 parameters 3 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H-atom parameters constrained
w = 1/[σ2(F
o2) + (0.0499P)2 + 0.2519P]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max < 0.001
Δρmax = 0.22 e Å−3
Δρmin = −0.20 e Å−3
Special details
supporting information
sup-2 Acta Cryst. (2005). E61, o4200–o4202
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2,
conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used
only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2
are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
C1 0.5030 (3) 0.62613 (14) 0.10178 (17) 0.0464 (4) H1 0.5094 0.6303 0.0115 0.056* C2 0.3533 (2) 0.67910 (13) 0.15543 (14) 0.0363 (3) C3 0.0983 (2) 0.77652 (13) 0.20040 (14) 0.0360 (3) C4 0.3419 (2) 0.67417 (13) 0.29114 (14) 0.0356 (3) C5 0.4837 (3) 0.61412 (14) 0.37693 (16) 0.0453 (4) H5 0.4783 0.6097 0.4673 0.054* C6 0.6325 (3) 0.56140 (16) 0.32379 (18) 0.0523 (4) H6 0.7290 0.5210 0.3795 0.063* C7 0.6415 (3) 0.56717 (15) 0.18836 (19) 0.0523 (4) H7 0.7437 0.5303 0.1558 0.063* C8 −0.1792 (3) 0.87225 (18) 0.3269 (2) 0.0639 (6) H8A −0.2960 0.9182 0.3220 0.096* H8B −0.0773 0.9068 0.3896 0.096* H8C −0.2045 0.7945 0.3550 0.096* N1 0.17943 (18) 0.73652 (11) 0.31710 (12) 0.0372 (3) N2 0.19456 (19) 0.74460 (12) 0.09984 (13) 0.0400 (3) S1 −0.10474 (6) 0.86698 (4) 0.16743 (4) 0.0520 (2) H2N 0.164 (3) 0.7588 (14) 0.018 (2) 0.047 (5)*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
C1 0.0557 (10) 0.0493 (9) 0.0383 (9) −0.0028 (7) 0.0196 (7) −0.0051 (7) C2 0.0429 (8) 0.0383 (8) 0.0287 (7) −0.0054 (6) 0.0089 (6) −0.0023 (6) C3 0.0365 (8) 0.0437 (8) 0.0277 (7) −0.0039 (6) 0.0046 (6) 0.0008 (6) C4 0.0412 (8) 0.0385 (8) 0.0276 (7) −0.0023 (6) 0.0073 (6) −0.0019 (6) C5 0.0521 (10) 0.0507 (9) 0.0322 (8) 0.0062 (7) 0.0040 (7) 0.0010 (7) C6 0.0516 (10) 0.0517 (10) 0.0521 (11) 0.0114 (8) 0.0032 (8) 0.0001 (8) C7 0.0516 (10) 0.0502 (10) 0.0590 (11) 0.0057 (8) 0.0205 (8) −0.0082 (8) C8 0.0615 (12) 0.0683 (13) 0.0684 (13) 0.0192 (9) 0.0302 (10) 0.0111 (10) N1 0.0399 (7) 0.0461 (7) 0.0258 (6) 0.0018 (5) 0.0063 (5) 0.0001 (5) N2 0.0461 (8) 0.0527 (8) 0.0209 (6) −0.0010 (6) 0.0049 (5) 0.0022 (5) S1 0.0425 (3) 0.0682 (3) 0.0443 (3) 0.00987 (19) 0.00372 (19) 0.01144 (19)
Geometric parameters (Å, º)
supporting information
sup-3 Acta Cryst. (2005). E61, o4200–o4202
C2—N2 1.378 (2) C6—H6 0.9300 C2—C4 1.400 (2) C7—H7 0.9300 C3—N1 1.3160 (19) C8—S1 1.783 (2) C3—N2 1.3584 (19) C8—H8B 0.9600 C3—S1 1.7374 (16) C8—H8A 0.9600 C4—C5 1.391 (2) C8—H8C 0.9600 C4—N1 1.3931 (19) N2—H2N 0.84 (2)
C7—C1—C2 116.86 (15) C5—C6—H6 119.3 C7—C1—H1 121.6 C7—C6—H6 119.3 C2—C1—H1 121.6 C1—C7—C6 121.59 (15) N2—C2—C1 132.41 (14) C1—C7—H7 119.2 N2—C2—C4 105.27 (13) C6—C7—H7 119.2 C1—C2—C4 122.32 (15) S1—C8—H8B 109.5 N1—C3—N2 113.48 (13) S1—C8—H8A 109.5 N1—C3—S1 126.90 (11) H8B—C8—H8A 109.5 N2—C3—S1 119.56 (11) S1—C8—H8C 109.5 C5—C4—N1 130.26 (13) H8B—C8—H8C 109.5 C5—C4—C2 119.85 (14) H8A—C8—H8C 109.5 N1—C4—C2 109.89 (13) C3—N1—C4 104.45 (12) C6—C5—C4 117.90 (15) C3—N2—C2 106.91 (12) C6—C5—H5 121.0 C3—N2—H2N 128.4 (12) C4—C5—H5 121.0 C2—N2—H2N 124.7 (12) C5—C6—C7 121.48 (16) C3—S1—C8 100.69 (8)
C7—C1—C2—N2 179.38 (16) N2—C3—N1—C4 0.35 (17) C7—C1—C2—C4 0.3 (2) S1—C3—N1—C4 −176.85 (12) N2—C2—C4—C5 −179.55 (14) C5—C4—N1—C3 179.07 (17) C1—C2—C4—C5 −0.2 (2) C2—C4—N1—C3 0.06 (16) N2—C2—C4—N1 −0.43 (16) N1—C3—N2—C2 −0.63 (18) C1—C2—C4—N1 178.88 (14) S1—C3—N2—C2 176.79 (10) N1—C4—C5—C6 −178.72 (16) C1—C2—N2—C3 −178.59 (16) C2—C4—C5—C6 0.2 (2) C4—C2—N2—C3 0.61 (16) C4—C5—C6—C7 −0.2 (3) N1—C3—S1—C8 −6.48 (16) C2—C1—C7—C6 −0.3 (3) N2—C3—S1—C8 176.47 (13) C5—C6—C7—C1 0.3 (3)
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
N2—H2N···N1i 0.84 (2) 2.07 (2) 2.8769 (17) 160.3 (16)
C8—H8A···Cg1ii 0.96 2.71 3.58 150