metal-organic papers
Acta Cryst.(2006). E62, m67–m68 doi:10.1107/S1600536805040857 Liuet al. [Zn(C
2H4NO2)2(C6H6N4S2)]2H2O
m67
Acta Crystallographica Section E Structure Reports
Online
ISSN 1600-5368
(2,2
000-Diamino-4,4
000-bi-1,3-thiazole-j
2N
,
N
000)-bis(glycinato-j
2N
,
O
)zinc(II) dihydrate
Bing-Xin Liu,aJian-Yong Yuaand Duan-Jun Xub*
a
Department of Chemistry, Shanghai University, People’s Republic of China, andbDepartment of Chemistry, Zhejiang University, People’s Republic of China
Correspondence e-mail: xudj@mail.hz.zj.cn
Key indicators
Single-crystal X-ray study
T= 295 K
Mean(C–C) = 0.002 A˚
Rfactor = 0.021
wRfactor = 0.054
Data-to-parameter ratio = 16.7
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2006 International Union of Crystallography Printed in Great Britain – all rights reserved
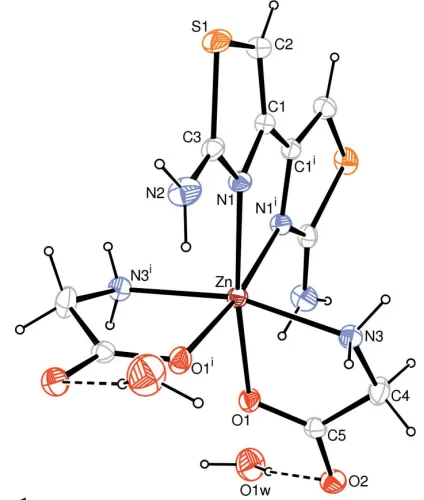
In the title ZnIIcomplex, [Zn(C2H4NO2)2(C6H6N4S2)]2H2O, the ZnII ion is coordinated by two glycinate anions and a diaminobithiazole (DABT) molecule in a distorted octahedral geometry. Two thiazole rings of the same DABT are twisted with respect to each other with a dihedral angle of 10.56 (6). The glycinate chelates to the ZnIIion by the amino N and carboxylate O atoms; the chelating five-membered ring displays an envelope configuration. A twofold rotation axis passes through the Zn atom and the mid-point of the C—C bond linking the two thiazole rings.
Comment
We are interested in metal complexes with diaminobithiazole (DABT) because of their potential magnetic properties (Sun et al., 1997). As part of an ongoing investigation of DABT complexes (Liu et al., 2001), we present here the crystal structure of the title ZnIIcomplex, (I).
The molecular structure of (I) is shown in Fig. 1. A twofold rotation axis passes through the Zn atom and the mid-point of the C—C bond linking the two thiazole rings. Two glycinate anions and one DABT molecule chelate to the ZnIIion in a distorted octahedral geometry (Table 1). Thiazole rings of the same DABT are twisted with respect to the each other with a dihedral angle of 10.56 (6), comparable to the CoII analog [9.68 (6); Yuet al., 2005]. The chelating five-membered ring of the glycinate anion displays an envelope conformation, atom N3 being displaced 0.390 (2) A˚ from the mean plane formed by the other four atoms.
The classical O—H O and N—H O hydrogen-bonding network stabilizes the crystal structure (Table 2).
Experimental
An aqueous solution (20 ml) containing DABT (1 mmol) and ZnCl2
(1 mmol) was mixed with another aqueous solution (10 ml) of glycine (2 mmol) and NaOH (1 mmol). The mixture was refluxed for 4 h. The
solution was filtered after cooling to room temperature. Single crys-tals of (I) were obtained from the filtrate after one week.
Crystal data
[Zn(C2H4NO2)2(C6H6N4S2)]2H2O Mr= 447.79
Monoclinic,C2=c a= 13.121 (2) A˚
b= 9.0063 (16) A˚
c= 14.124 (2) A˚
= 92.365 (6)
V= 1667.7 (5) A˚3
Z= 4
Dx= 1.783 Mg m
3
MoKradiation Cell parameters from 5886
reflections
= 2.8–25.0
= 1.77 mm1
T= 295 (2) K Prism, colorless 0.250.200.16 mm
Data collection
Rigaku R-AXIS RAPID diffractometer
!scans
Absorption correction: multi-scan (ABSCOR; Higashi, 1995)
Tmin= 0.628,Tmax= 0.750
7912 measured reflections
1918 independent reflections 1808 reflections withI> 2(I)
Rint= 0.018
max= 27.5
h=16!16
k=10!11
l=18!18
Refinement
Refinement onF2 R[F2> 2(F2)] = 0.021
wR(F2) = 0.054 S= 1.08 1918 reflections 115 parameters
H-atom parameters constrained
w= 1/[2(F
o2) + (0.0209P)2
+ 2.0364P]
whereP= (Fo2+ 2Fc2)/3
(/)max= 0.001 max= 0.30 e A˚
3
min=0.22 e A˚
3
Extinction correction:SHELXL97
Extinction coefficient: 0.0134 (5)
Table 1
Selected bond lengths (A˚ ).
Zn—O1 2.1714 (11)
Zn—N1 2.1823 (13)
Zn—N3 2.1185 (13)
S1—C2 1.7219 (19)
S1—C3 1.7417 (17)
N1—C1 1.3901 (19)
N1—C3 1.3212 (19)
N2—C3 1.333 (2)
C1—C2 1.344 (2)
C1—C1i
1.469 (3)
Symmetry code: (i)xþ1;y;zþ1 2.
Table 2
Hydrogen-bond geometry (A˚ ,).
D—H A D—H H A D A D—H A
O1W—H1A O1ii
0.88 2.18 2.976 (2) 151
O1W—H1B O2 0.90 2.00 2.878 (2) 167
N2—H2A O1i
0.87 2.37 3.098 (2) 141
N2—H2B O2iii
0.86 2.09 2.9059 (19) 159
N3—H3A O2iv
0.88 2.47 3.3209 (19) 165
N3—H3B O1Wv
0.90 2.22 3.051 (2) 154
Symmetry codes: (i) xþ1;y;zþ1
2; (ii) xþ1;yþ2;zþ1; (iii)
x1 2;yþ
3 2;z
1 2; (iv)xþ
3 2;y
1 2;zþ
1
2; (v)x;yþ2;z 1 2.
H atoms bonded to C atoms were placed in calculated positions, with C—H = 0.93 A˚ (aromatic) or 0.97 A˚ (methylene), and included in the final cycles of refinement as riding,withUiso(H) = 1.2Ueq(C).
Other H atoms were located in a difference Fourier map and refined as riding in their as-found relative positions with Uiso(H) =
1.5Ueq(carrier).
Data collection: PROCESS-AUTO (Rigaku, 1998); cell refine-ment:PROCESS-AUTO; data reduction:CrystalStructure(Rigaku/ MSC, 2002); program(s) used to solve structure:SIR92(Altomareet al., 1993); program(s) used to refine structure: SHELXL97 (Shel-drick, 1997); molecular graphics: ORTEP3 for Windows(Farrugia, 1997); software used to prepare material for publication: WinGX
(Farrugia, 1999).
The project was supported by the Educational Develop-ment Foundation of Shanghai Educational Committee, China (No. AB0448).
References
Altomare, A., Cascarano, G., Giacovazzo, C. & Guagliardi, A. (1993).J. Appl. Cryst.26, 343–350.
Farrugia, L. J. (1997).J. Appl. Cryst.30, 565. Farrugia, L. J. (1999).J. Appl. Cryst.32, 837–838.
Higashi, T. (1995).ABSCOR. Rigaku Corporation, Tokyo, Japan.
Liu, J.-G., Nie, J.-J., Xu, D.-J., Xu, Y.-Z., Wu, J.-Y. & Chiang, M. Y. (2001).Acta Cryst.C57, 354–355.
Rigaku (1998).PROCESS-AUTO. Rigaku Corporation, Tokyo, Japan. Rigaku/MSC (2002).CrystalStructure. Version 3.00. Rigaku/MSC, 900 New
Trails Drive, The Woodlands, TX 77381-5209, USA.
[image:2.610.332.544.74.324.2]Sheldrick, G. M. (1997).SHELXL97. University of Go¨ttingen, Germany. Sun, W., Gao, X. & Lu, F.-J. (1997).Appl. Polym. Sci.64, 2309–2315. Yu, J.-R., Liu, B.-X. & Xu, D.-J. (2005).Acta Cryst.E61, m2232–m2233.
Figure 1
The molecular structure of (I), with 30% probability displacement ellipsoids (arbitrary spheres for H atoms). Dashed lines indicate hydrogen bonds [symmetry code: (i)x+ 1,y,z+1
supporting information
sup-1
Acta Cryst. (2006). E62, m67–m68
supporting information
Acta Cryst. (2006). E62, m67–m68 [doi:10.1107/S1600536805040857]
(2,2
′
-Diamino-4,4
′
-bi-1,3-thiazole-
κ
2N
,
N
′
)bis(glycinato-
κ
2N
,
O
)zinc(II)
dihydrate
Bing-Xin Liu, Jian-Yong Yu and Duan-Jun Xu
S1. Comment
We are interested in metal complexes with diaminobithiazole (DABT) because of their potential magnetic properties (Sun
et al., 1997). As part of an ongoing investigation of DABT complexes (Liu et al., 2001), we present here the X-ray
structure of the title ZnII complex, (I).
The molecular structure of (I) is shown in Fig. 1. The ZnII complex has twofold symmetry. Two glycinate anions and
one DABT molecule chelate to the ZnII ion in a distorted octahedral geometry (Table 1). Thiazole rings of the same
DABT are twisted with respect to the each other with a dihedral angle of 10.56 (6)°, comparable to the CoII analogue
[9.68 (6)°; Yu et al., 2005]. The chelating five-membered ring of the glycinate anion displays an envelope configuration,
the N3 atom being displaced 0.390 (2) Å from the mean plane formed by other four atoms.
The classic O—H···O and N—H···O hydrogen-bonding network stabilizes the crystal structure (Table 2).
S2. Experimental
An aqueous solution (20 ml) containing DABT (1 mmol) and ZnCl2 (1 mmol) was mixed with another aqueous solution
(10 ml) of glycine (2 mmol) and NaOH (1 mmol). The mixture was refluxed for 4 h. The solution was filtered after
cooling to room temperature. Single crystals of (I) were obtained from the filtrate after one week.
S3. Refinement
H atoms bonded to C atoms were placed in calculated positions, with C—H = 0.93 Å (aromatic) or 0.97 Å (methylene),
and included in the final cycles of refinement as riding,with Uiso(H) = 1.2Ueq(C). Other H atoms were located in a
Figure 1
The molecular structure of (I), with 30% probability displacement ellipsoids (arbitrary spheres for H atoms). Dashed lines
indicate hydrogen bonds [symmetry code: (i) −x + 1,y,-z + 1/2].
(2,2′-Diamino-4,4′-bi-1,3-thiazole-κ2N,N′)bis(glycinato-κ2N,O)zinc(II) dihydrate
Crystal data
[Zn(C2H4NO2)2(C6H6N4S2)]·2H2O Mr = 447.79
Monoclinic, C2/c Hall symbol: -C 2yc a = 13.121 (2) Å b = 9.0063 (16) Å c = 14.124 (2) Å β = 92.365 (6)°
V = 1667.7 (5) Å3 Z = 4
F(000) = 920 Dx = 1.783 Mg m−3
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 5886 reflections θ = 2.8–25.0°
supporting information
sup-3
Acta Cryst. (2006). E62, m67–m68
T = 295 K Prism, colorless
0.25 × 0.20 × 0.16 mm
Data collection
Rigaku R-AXIS RAPID diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
Detector resolution: 10 pixels mm-1 ω scans
Absorption correction: multi-scan (ABSCOR; Higashi, 1995) Tmin = 0.628, Tmax = 0.750
7912 measured reflections 1918 independent reflections 1808 reflections with I > 2σ(I) Rint = 0.018
θmax = 27.5°, θmin = 2.7° h = −16→16
k = −10→11 l = −18→18
Refinement
Refinement on F2
Least-squares matrix: full R[F2 > 2σ(F2)] = 0.021 wR(F2) = 0.054 S = 1.08 1918 reflections 115 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H-atom parameters constrained w = 1/[σ2(F
o2) + (0.0209P)2 + 2.0364P]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max = 0.001
Δρmax = 0.30 e Å−3
Δρmin = −0.22 e Å−3
Extinction correction: SHELXL97, Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4
Extinction coefficient: 0.0134 (5)
Special details
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2,
conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used
only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2
are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
Zn 0.5000 0.71733 (3) 0.2500 0.02228 (10)
S1 0.37183 (3) 0.32835 (5) 0.05293 (3) 0.03481 (12) N1 0.45516 (10) 0.52922 (14) 0.16024 (9) 0.0238 (3) N2 0.37698 (11) 0.62134 (18) 0.01891 (10) 0.0373 (3)
H2A 0.3961 0.7127 0.0285 0.056*
H2B 0.3357 0.6014 −0.0281 0.056*
N3 0.64785 (10) 0.74766 (16) 0.19830 (9) 0.0271 (3)
H3A 0.6760 0.6673 0.1761 0.041*
H3B 0.6445 0.8100 0.1486 0.041*
H1A 0.5571 0.9931 0.5852 0.090*
H1B 0.6401 0.9832 0.5208 0.090*
C1 0.47278 (11) 0.39244 (17) 0.20349 (10) 0.0244 (3) C2 0.43515 (13) 0.27356 (18) 0.15629 (12) 0.0318 (3)
H2 0.4423 0.1757 0.1766 0.038*
C3 0.40309 (12) 0.51226 (18) 0.07889 (11) 0.0265 (3) C4 0.71545 (13) 0.8164 (2) 0.27111 (13) 0.0360 (4)
H4A 0.7625 0.8824 0.2407 0.043*
H4B 0.7552 0.7394 0.3034 0.043*
C5 0.65800 (11) 0.90340 (17) 0.34356 (10) 0.0250 (3)
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
Zn 0.02056 (14) 0.02147 (15) 0.02446 (14) 0.000 −0.00306 (9) 0.000
S1 0.0346 (2) 0.0353 (2) 0.0343 (2) −0.00850 (17) −0.00265 (16) −0.01341 (17) N1 0.0244 (6) 0.0231 (6) 0.0234 (6) −0.0012 (5) −0.0036 (5) −0.0025 (5) N2 0.0414 (8) 0.0401 (8) 0.0292 (7) −0.0059 (7) −0.0133 (6) 0.0006 (6) N3 0.0275 (7) 0.0290 (7) 0.0248 (6) −0.0001 (5) 0.0011 (5) −0.0038 (5) O1 0.0235 (5) 0.0321 (6) 0.0344 (6) −0.0006 (5) 0.0007 (4) −0.0096 (5) O2 0.0332 (6) 0.0327 (6) 0.0357 (6) −0.0029 (5) −0.0096 (5) −0.0071 (5) O1W 0.0585 (10) 0.0659 (11) 0.0558 (9) 0.0021 (8) 0.0156 (8) −0.0077 (8) C1 0.0207 (7) 0.0235 (7) 0.0292 (8) −0.0002 (6) 0.0023 (6) −0.0023 (6) C2 0.0314 (8) 0.0254 (8) 0.0388 (9) −0.0030 (6) 0.0023 (7) −0.0046 (7) C3 0.0226 (7) 0.0307 (8) 0.0261 (7) −0.0033 (6) 0.0000 (6) −0.0064 (6) C4 0.0223 (7) 0.0468 (10) 0.0388 (9) −0.0021 (7) 0.0008 (6) −0.0136 (8) C5 0.0262 (7) 0.0224 (7) 0.0259 (7) 0.0011 (6) −0.0046 (6) 0.0014 (5)
Geometric parameters (Å, º)
Zn—O1 2.1714 (11) N3—C4 1.467 (2)
Zn—O1i 2.1714 (11) N3—H3A 0.8765
Zn—N1 2.1823 (13) N3—H3B 0.8990
Zn—N1i 2.1823 (13) O1—C5 1.2629 (19)
Zn—N3i 2.1185 (13) O2—C5 1.2516 (19)
Zn—N3 2.1185 (13) O1W—H1A 0.8763
S1—C2 1.7219 (19) O1W—H1B 0.8952
S1—C3 1.7417 (17) C1—C2 1.344 (2)
N1—C1 1.3901 (19) C1—C1i 1.469 (3)
N1—C3 1.3212 (19) C2—H2 0.9300
N2—C3 1.333 (2) C4—C5 1.514 (2)
N2—H2A 0.8692 C4—H4A 0.9700
N2—H2B 0.8585 C4—H4B 0.9700
N3i—Zn—N3 165.18 (8) Zn—N3—H3A 115.0
N3i—Zn—O1 91.32 (5) C4—N3—H3B 107.0
N3—Zn—O1 78.46 (5) Zn—N3—H3B 109.5
supporting information
sup-5
Acta Cryst. (2006). E62, m67–m68
N3—Zn—O1i 91.32 (5) C5—O1—Zn 114.99 (10)
O1—Zn—O1i 93.25 (7) H1A—O1W—H1B 109.9
N3i—Zn—N1 94.11 (5) C2—C1—N1 115.90 (14)
N3—Zn—N1 97.39 (5) C2—C1—C1i 126.76 (10)
O1—Zn—N1 171.33 (5) N1—C1—C1i 117.31 (8)
O1i—Zn—N1 94.46 (5) C1—C2—S1 110.28 (13)
N3i—Zn—N1i 97.39 (5) C1—C2—H2 124.9
N3—Zn—N1i 94.11 (5) S1—C2—H2 124.9
O1—Zn—N1i 94.46 (5) N1—C3—N2 125.29 (15)
O1i—Zn—N1i 171.33 (5) N1—C3—S1 113.69 (12)
N1—Zn—N1i 78.15 (7) N2—C3—S1 121.00 (12)
C2—S1—C3 89.59 (8) N3—C4—C5 112.87 (13)
C3—N1—C1 110.51 (13) N3—C4—H4A 109.0
C3—N1—Zn 135.43 (11) C5—C4—H4A 109.0
C1—N1—Zn 113.37 (9) N3—C4—H4B 109.0
C3—N2—H2A 122.3 C5—C4—H4B 109.0
C3—N2—H2B 118.4 H4A—C4—H4B 107.8
H2A—N2—H2B 119.2 O2—C5—O1 124.57 (15)
C4—N3—Zn 110.40 (10) O2—C5—C4 117.07 (14)
C4—N3—H3A 110.4 O1—C5—C4 118.32 (14)
Symmetry code: (i) −x+1, y, −z+1/2.
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
O1W—H1A···O1ii 0.88 2.18 2.976 (2) 151
O1W—H1B···O2 0.90 2.00 2.878 (2) 167
N2—H2A···O1i 0.87 2.37 3.098 (2) 141
N2—H2B···O2iii 0.86 2.09 2.9059 (19) 159
N3—H3A···O2iv 0.88 2.47 3.3209 (19) 165
N3—H3B···O1Wv 0.90 2.22 3.051 (2) 154