organic papers
Acta Cryst.(2006). E62, o1957–o1958 doi:10.1107/S1600536806013560 Slateret al. C10H11NO2
o1957
Acta Crystallographica Section EStructure Reports Online
ISSN 1600-5368
N
-(2-Acetylphenyl)acetamide
Heather L. Slater, Hanna Rozynski, Guy Crundwell and Neil M. Glagovich*
Department of Chemistry, Central Connecticut State University, New Britain, CT 06053, USA
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study T= 298 K
Mean(C–C) = 0.003 A˚ Rfactor = 0.048 wRfactor = 0.118
Data-to-parameter ratio = 13.2
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
Received 24 March 2006 Accepted 13 April 2006
#2006 International Union of Crystallography All rights reserved
The title compound, C10H11NO2, was synthesized from 20
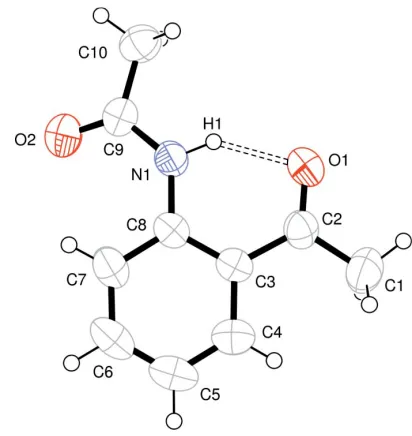
-aminoacetophenone in acetic anhydride. In the molecular structure, an intramolecular N—H O hydrogen bond [H O = 1.893 (18) A˚ ] appears to affect the overall planar conformation of the molecule.
Comment
Derivatives of acetophenone have been synthesized for many reasons: as precursors of indoles (Fuerstner et al., 1991; Fuerstner & Jumbam, 1992) and quinolines (Curran & Kuo, 1984), in order to study their chemiluminescent properties (Pileni & Santus, 1977, Giraud et al., 1977; Sugiyama & Akutagawa, 1967), as potential analgesic precursors (Giuliani et al., 1983; Lemboet al., 1983), and to study intramolecular hydrogen bonding (Appleton et al., 1970; Hambly & Bonnyman, 1958). Our special interest in acetophenone derivatives results from their use in the synthesis of unsym-metrical Tro¨ger’s base analogs (Webb & Wilcox, 1990; Pardoet al., 2001; Jensenet al., 2002).
The title compound, (I) (Fig. 1), was synthesized as an intermediate in the total synthesis of 6-methyl-2-nitro-6H,12H-5,11-methanodibenzo[b,f][1,5]diazocine, an unsym-metrical Tro¨ger’s base compound. A key step in the synthesis involved Schiff base formation between the acetyl O atom of compound (I) and the amino N atom of p-nitroaniline. The imine product did not form, although several methodologies were attempted (Weingartenet al., 1967). It is possible that the intramolecular hydrogen bond between the amide H atom and acetyl O atom somehow interferes with the condensation reaction.
six-membered ring intramolecular hydrogen bonds [two recent examples have been reported by Manhet al.(1999) and Ando et al.(2004)]. The molecule of (I) is essentially planar, with an r.m.s. deviation of 0.0420 A˚ for atoms O1/N1/C1–C8, while atoms C9, C10 and O2 are displaced by 0.344 (2), 0.321 (3) and 0.645 (2) A˚ , respectively, from this plane.
Experimental
The title compound, C10H11NO2, was synthesized according to a
previously reported method (Leonard & Boyd, 1946). 20
-Amino-acetophenone (5 g, 37 mmol) was dissolved in acetic anhydride (10 ml) and stirred at room temperature for 3 h. The resulting clear solution was poured on to crushed ice (100 ml) and allowed to stand until all of the excess acetic anhydride had been hydrolyzed. The white precipitate which formed was filtered off and recrystallized from ethanol to yield 6.3 g (96%) ofN-(2-acetylphenyl)acetamide.
Crystal data
C10H11NO2
Mr= 177.20
Monoclinic,P21=c
a= 7.765 (7) A˚
b= 8.699 (7) A˚
c= 15.805 (13) A˚
= 119.35 (7)
V= 930.6 (14) A˚3
Z= 4
Dx= 1.265 Mg m
3
MoKradiation
= 0.09 mm1
T= 298 (2) K Needle, orange 0.430.310.27 mm
Data collection
Oxford Diffraction Sapphire3 diffractometer
!scans
Absorption correction: multi-scan (CrysAlis RED; Oxford Diffraction, 2005)’
19878 measured reflections 1639 independent reflections 1019 reflections withI> 2(I)
Rint= 0.036
max= 25.0
Refinement
Refinement onF2 R[F2> 2(F2)] = 0.048
wR(F2) = 0.118
S= 0.93 1639 reflections 124 parameters
H atoms treated by a mixture of independent and constrained refinement
w= 1/[2(F
o2) + (0.0807P)2]
whereP= (Fo2+ 2Fc2)/3
(/)max< 0.001 max= 0.18 e A˚
3 min=0.26 e A˚
3
Table 1
Hydrogen-bond geometry (A˚ ,).
D—H A D—H H A D A D—H A
N1—H1 O1 0.888 (17) 1.893 (18) 2.657 (2) 143.1 (15)
H atoms bonded to C atoms were placed in calculated postions, with C—H = 0.93 A˚ , or 0.96 A˚ for methyl groups, and included in the refinement in a riding-model approximation, with Uiso(H) =
1.2Ueq(C), or 1.5Ueq(C) for methyl H atoms. The N-bound H atom
was refined independently with an isotropic displacement parameter. Data collection: CrysAlis CCD (Oxford Diffraction, 2005); cell refinement: CrysAlis RED (Oxford Diffraction, 2005); data reduc-tion:CrysAlis RED; program(s) used to solve structure:SHELXS97
(Sheldrick, 1997); program(s) used to refine structure:SHELXL97
(Sheldrick, 1997); molecular graphics: ORTEP-3 (Farrugia, 1997); software used to prepare material for publication:SHELXL97.
This research was funded in part by an NIH Area Grant (No. 1 R15 AI057408–01) and an NSF MRI Grant (No. 0520982), and also by CCSU-AAUP research grants and CCSU Faculty Student research grants. GC acknowledges the NSF (MRI Grant No. 0420322).
References
Ando, K., Tsuji, E., Ando, Y., Kuwata, N., Kunitomo, J., Yamashita, M., Ohta, S., Kohno, S. & Ohishi, Y. (2004).Org. Biomol. Chem.2, 625–635. Appleton, J. M., Andrews, B. D., Rae, I. D. & Reichert, B. E. (1970).Aust. J.
Chem.23, 1667–1677.
Curran, D. P. & Kuo, S. C. (1984).J. Org. Chem.49, 2063–2065. Farrugia, L. J. (1997).J. Appl. Cryst.30, 565.
Fuerstner, A. & Jumbam, D. N. (1992).Tetrahedron,48, 5991–6010. Fuerstner, A., Jumbam, D. N. & Weidmann, H. (1991).Tetrahedron Lett.32,
6695–6696.
Giraud, M., Pileni, M. P., Valla, A. & Santus, R. (1977).J. Chim. Phys. Phys. Chim. Biol.74, 224–228.
Giuliani, E., Lembo, S., Sasso, V., Sorrentino, L., Silipo, C. & Vittoria, A. (1983).Farmaco Ed. Sci.38, 847–864.
Hambly, A. N. & Bonnyman, J. (1958).Aust. J. Chem.11, 529–537. Jensen, J., Tejler, J. & Wa¨rnmark, K. (2002).J. Org. Chem.67, 6008–6014. Lembo, S., Sasso, V., Silipo, C. & Vittoria, A. (1983).Farmaco Ed. Sci.38, 750–
761.
Leonard, N. J. & Boyd, S. N. Jr (1946).J. Org. Chem.11, 405–418.
Manh, G. T., Purseigle, F., Dubreuil, D., Predere, J. P., Guingant, A., Danion-Bougot, R. & Toupet, L. (1999).J. Chem. Soc. Perkin Trans. 1, pp. 2821– 2828.
Oxford Diffraction (2005). CrysAlis CCD and CrysAlis RED. Versions 1.171.27p5 beta. Oxford Diffraction Ltd, Abingdon, Oxfordshire, England. Pardo, C., Sesmilo, E., Gutie´rrez-Puebla, E., Monge, A., Elguero, J. &
Fruchier, A. (2001).J. Org. Chem.66, 1607–1611. Pileni, M. P. & Santus, R. (1977).J. Phys. Chem.81, 755–760.
Sheldrick, G. M. (1997). SHELXL97 and SHELXS97. University of Go¨ttingen, Germany.
Sugiyama, N. & Akutagawa, M. (1967).Bull. Chem. Soc. Jpn,40, 240–244. Webb, T. H. & Wilcox, C. S. (1990).J. Org. Chem.55, 363–365.
[image:2.610.64.270.70.288.2]Weingarten, H., Chupp, J. P. & White, W. A. (1967).J. Org. Chem. , 3246–
Figure 1
supporting information
sup-1 Acta Cryst. (2006). E62, o1957–o1958
supporting information
Acta Cryst. (2006). E62, o1957–o1958 [https://doi.org/10.1107/S1600536806013560]
N
-(2-Acetylphenyl)acetamide
Heather L. Slater, Hanna Rozynski, Guy Crundwell and Neil M. Glagovich
N-(2-acetylphenyl)acetamide
Crystal data
C10H11NO2
Mr = 177.20
Monoclinic, P21/c
Hall symbol: -P 2ybc
a = 7.765 (7) Å
b = 8.699 (7) Å
c = 15.805 (13) Å
β = 119.35 (7)°
V = 930.6 (14) Å3
Z = 4
F(000) = 376
Dx = 1.265 Mg m−3
Melting point: 349 K
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 5840 reflections
θ = 3.8–32.9°
µ = 0.09 mm−1
T = 298 K Needle, orange 0.43 × 0.31 × 0.27 mm
Data collection
Oxford Diffraction Sapphire3 diffractometer
Radiation source: Enhance (Mo) X-ray Source Graphite monochromator
Detector resolution: 16.1790 pixels mm-1
ω scans
Absorption correction: multi-scan
(CrysAlis RED; Oxford Diffraction, 2005)′
Tmin = 0.809, Tmax = 0.975
19878 measured reflections 1639 independent reflections 1019 reflections with I > 2σ(I)
Rint = 0.036
θmax = 25.0°, θmin = 3.8°
h = −9→9
k = −10→10
l = −18→18
Refinement
Refinement on F2
Least-squares matrix: full
R[F2 > 2σ(F2)] = 0.048
wR(F2) = 0.118
S = 0.93 1639 reflections 124 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H atoms treated by a mixture of independent and constrained refinement
w = 1/[σ2(F
o2) + (0.0807P)2]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max < 0.001
Δρmax = 0.18 e Å−3
Special details
Experimental. Spectroscopic analysis: Rf = 0.46 (Al2O3, 80% hexanes/20% ethyl acetate); m.p. 349 K; IR (nujol, ν,
cm-1): 3222, 3065, 1687, 1652, 1584, 1529, 1454, 1251, 765, 723; 1H NMR (400 MHz, CDCl
3, δ, p.p.m.): 11.706 (s, 1H),
8.735 (dd, 1H, J = 8.3 and 0.9 Hz), 7.888 (dd, 1H, J = 7.8 and 1.4 Hz), 7.546 (dt, 1H, J = 8.3 and 1.4 Hz), 7.104 (dt, 1H, J = 7.8 and 0.9 Hz), 2.661 (s, 3H), 2.222 (s, 3H); 13C NMR (400 MHz, CDCl
3, δ, p.p.m.): 202.94, 169.96, 141.09, 135.26,
131.67, 122.39, 121.78, 120.83, 28.72, 25.69; UV–Vis (CH2Cl2; λmax, logε): 326 nm, 3.72; EI–MS calculated for
C10H11NO2: M+ 177; found: 177.
During model refinement, H atoms bonded to C atoms were placed in calculated postions with C—H = 0.93 or C—H = 0.96 Å (for methyl groups) and included in the refinement in a riding-model approximation with Uiso(H) = either
1.2Ueq(C) or 1.5Ueq(C) for methyl H atoms. The H atom bonded to N was refined independently with an isotropic
displacement parameter.
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2,
conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used
only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2
are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
supporting information
sup-3 Acta Cryst. (2006). E62, o1957–o1958
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
O1 0.0757 (8) 0.0732 (9) 0.0541 (7) 0.0048 (6) 0.0124 (6) −0.0073 (6) C2 0.0643 (11) 0.0505 (10) 0.0561 (10) −0.0087 (8) 0.0308 (9) −0.0010 (8) C1 0.1073 (16) 0.0580 (12) 0.0802 (13) −0.0036 (11) 0.0415 (12) −0.0104 (10) C3 0.0458 (9) 0.0539 (10) 0.0489 (9) −0.0031 (7) 0.0241 (7) 0.0052 (7) C4 0.0600 (10) 0.0667 (12) 0.0662 (11) 0.0067 (9) 0.0326 (9) 0.0126 (9) C5 0.0542 (11) 0.0897 (15) 0.0648 (12) 0.0091 (10) 0.0203 (9) 0.0228 (11) C6 0.0625 (12) 0.0962 (16) 0.0466 (9) −0.0153 (11) 0.0129 (9) 0.0056 (10) C7 0.0659 (11) 0.0677 (12) 0.0480 (9) −0.0113 (9) 0.0224 (8) −0.0037 (8) C8 0.0441 (8) 0.0555 (10) 0.0472 (8) −0.0071 (7) 0.0226 (7) 0.0034 (7) N1 0.0562 (8) 0.0490 (9) 0.0459 (8) −0.0035 (6) 0.0210 (7) −0.0009 (6) C9 0.0623 (10) 0.0544 (11) 0.0608 (10) −0.0014 (8) 0.0328 (9) −0.0051 (9) O2 0.1082 (11) 0.0686 (9) 0.0853 (10) 0.0103 (8) 0.0171 (8) −0.0232 (8) C10 0.0752 (12) 0.0581 (11) 0.0718 (11) 0.0057 (9) 0.0387 (10) 0.0094 (9)
Geometric parameters (Å, º)
O1—C2 1.232 (2) C6—C7 1.385 (3) C2—C3 1.492 (3) C6—H6 0.9300 C2—C1 1.500 (3) C7—C8 1.398 (3) C1—H1A 0.9600 C7—H7 0.9300 C1—H1B 0.9600 C8—N1 1.408 (2) C1—H1C 0.9600 N1—C9 1.352 (2) C3—C4 1.409 (2) N1—H1 0.888 (17) C3—C8 1.419 (2) C9—O2 1.220 (2) C4—C5 1.373 (3) C9—C10 1.506 (3) C4—H4 0.9300 C10—H10A 0.9600 C5—C6 1.375 (3) C10—H10B 0.9600 C5—H5 0.9300 C10—H10C 0.9600
C4—C5—H5 120.5 C9—C10—H10C 109.5 C6—C5—H5 120.5 H10A—C10—H10C 109.5 C5—C6—C7 121.19 (17) H10B—C10—H10C 109.5 C5—C6—H6 119.4
O1—C2—C3—C4 −175.63 (15) C6—C7—C8—C3 −1.3 (2) C1—C2—C3—C4 4.4 (2) C4—C3—C8—C7 2.4 (2) O1—C2—C3—C8 3.1 (2) C2—C3—C8—C7 −176.39 (14) C1—C2—C3—C8 −176.83 (14) C4—C3—C8—N1 −179.00 (13) C8—C3—C4—C5 −1.5 (2) C2—C3—C8—N1 2.3 (2) C2—C3—C4—C5 177.30 (14) C7—C8—N1—C9 −16.6 (2) C3—C4—C5—C6 −0.5 (3) C3—C8—N1—C9 164.75 (14) C4—C5—C6—C7 1.6 (3) C8—N1—C9—O2 0.0 (3) C5—C6—C7—C8 −0.7 (3) C8—N1—C9—C10 −177.64 (14) C6—C7—C8—N1 −179.94 (13)
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A