organic papers
Acta Cryst.(2007). E63, o191–o192 doi:10.1107/S1600536806051828 Lvet al. C
6H9NO3
o191
Acta Crystallographica Section E Structure Reports
Online
ISSN 1600-5368
N-Acryloyl-
L-alanine
Zhi-Fang Lv,aWen-Yuan Wu,a Rong Zhangband Jin-Tang Wanga*
aDepartment of Applied Chemistry, College of
Science, Nanjing University of Technology, Nanjing 210009, People’s Republic of China, andbCollege of Science, Nanjing University of Technology, Nanjing 210009, People’s Republic of China
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study
T= 294 K
Mean(C–C) = 0.003 A˚
Rfactor = 0.034
wRfactor = 0.082 Data-to-parameter ratio = 9.7
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
Received 28 November 2006 Accepted 30 November 2006
#2007 International Union of Crystallography
All rights reserved
The title compound, C6H9NO3, was prepared by a nucleophilic
substitution reaction of acryloyl chloride withl-alanine. In the
crystal structure, intermolecular N—H O and O—H O hydrogen bonds link the molecules into a three-dimensional network, in which they may be effective in the stabilization of the crystal structure.
Comment
The title compound, (I), is an important intermediate and also a free radical addition monomer for the syntheses of radia-tion-sensitive (Heilmann & Palensky, 1981), hydropholic (Heilmann & Rasmussen, 1984) and pressure-sensitive (Heilmann, 1979) polymers. The crystal structure determina-tion of (I) has been carried out in order to elucidate the molecular conformation. We report here the synthesis and the crystal structure of (I).
In the molecule of (I) (Fig. 1), the bond lengths and angles are within normal ranges (Allenet al., 1987). Atoms C2, C3, O2, N1 and C4 are nearly coplanar, with a dihedral angle of 1.2 (3) between the C2/C3/O2 and C3/N1/C4 planes.
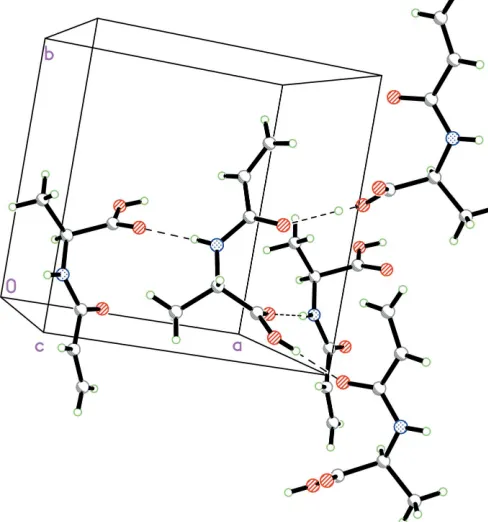
As can be seen from the packing diagram (Fig. 2),
inter-molecular N—H O and O—H O hydrogen bonds
(Table 1) link the molecules into a three-dimensional network, in which they may be effective in the stabilization of the crystal structure. Dipole–dipole and van der Waals interactions are also effective in the molecular packing.
Experimental
Crystal data
C6H9NO3
Mr= 143.14
Orthorhombic,P212121
a= 8.3670 (17) A˚
b= 8.7730 (18) A˚
c= 10.350 (2) A˚
V= 759.7 (3) A˚3
Z= 4
Dx= 1.251 Mg m 3
MoKradiation
= 0.10 mm1
T= 294 (2) K Block, colorless 0.400.300.30 mm
Data collection
Enraf–Nonius CAD-4 diffractometer
!/2scans
Absorption correction: scan (Northet al., 1968)
Tmin= 0.962,Tmax= 0.972
1686 measured reflections
888 independent reflections 805 reflections withI> 2(I)
Rint= 0.032 max= 26.0
3 standard reflections every 200 reflections intensity decay: none
Refinement
Refinement onF2
R[F2> 2(F2)] = 0.034
wR(F2) = 0.082
S= 1.08 888 reflections 92 parameters
H-atom parameters constrained
w= 1/[2(F
o2) + (0.04P)2
+ 0.05P]
whereP= (Fo2+ 2Fc2)/3
(/)max< 0.001
max= 0.18 e A˚3
min=0.13 e A˚3
Extinction correction:SHELXL97
Extinction coefficient: 0.317 (19)
Table 1
Hydrogen-bond geometry (A˚ ,).
D—H A D—H H A D A D—H A
N1—H1 O1i 0.86 2.01 2.861 (2) 171 O3—H3 O2ii
0.82 1.80 2.616 (2) 175
Symmetry codes: (i)xþ1 2;yþ
3
2;zþ2; (ii)x;y 1 2;zþ
3 2.
H atoms were positioned geometrically, with O—H = 0.82 A˚ , N— H = 0.86 A˚ and C—H = 0.93, 0.98, 0.93 and 0.96 A˚ for aromatic, methine, methylene and methyl H, respectively, and constrained to ride on their parent atoms, withUiso(H) =xUeq(C,N,O), wherex= 1.5
for OH and methyl H, and x = 1.2 for all other H atoms. In the absence of significant anomalous scattering effects, Friedel pairs were merged. The absolute configuration is known from the synthesis.
Data collection: CAD-4 Software (Enraf–Nonius, 1985); cell refinement: CAD-4 Software; data reduction: XCAD4 (Harms & Wocadlo, 1995); program(s) used to solve structure: SHELXS97
(Sheldrick, 1997); program(s) used to refine structure:SHELXL97
(Sheldrick, 1997); molecular graphics: SHELXTL (Bruker, 2000); software used to prepare material for publication:SHELXTL.
The authors thank the Center of Testing and Analysis, Nanjing University, for support.
References
Allen, F. H., Kennard, O., Watson, D. G., Brammer, L., Orpen, A. G. & Taylor,
Enraf–Nonius (1985).CAD-4 Software. Version 5.0. Enraf–Nonius, Delft, The Netherlands.
Harms, K. & Wocadlo, S. (1995).XCAD4. University of Marburg, Germany. Heilmann, S. M. (1979). US Patent No. 4 157 418.
Heilmann, S. M. & Palensky, F. J. (1981). US Patent No. 4 304 705. Heilmann, S. M. & Rasmussen, J. K. (1984). US Patent No. 4 451 619. North, A. C. T., Phillips, D. C. & Mathews, F. S. (1968).Acta Cryst.A24, 351–
[image:2.610.311.563.72.261.2]359. Figure 1
[image:2.610.315.559.304.565.2]The molecular structure of (I), showing the atom-numbering scheme. Displacement ellipsoids are drawn at the 30% probability level.
Figure 2
supporting information
sup-1 Acta Cryst. (2007). E63, o191–o192
supporting information
Acta Cryst. (2007). E63, o191–o192 [https://doi.org/10.1107/S1600536806051828]
N
-Acryloyl-
L-alanine
Zhi-Fang Lv, Wen-Yuan Wu, Rong Zhang and Jin-Tang Wang
N-acryloyl-L-alanine
Crystal data
C6H9NO3
Mr = 143.14
Orthorhombic, P212121
Hall symbol: P 2ac 2ab
a = 8.3670 (17) Å
b = 8.7730 (18) Å
c = 10.350 (2) Å
V = 759.7 (3) Å3
Z = 4
F(000) = 304
Dx = 1.251 Mg m−3
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 25 reflections
θ = 11–13°
µ = 0.10 mm−1
T = 294 K Block, colorless 0.40 × 0.30 × 0.30 mm
Data collection
Enraf–Nonius CAD-4 diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
ω/2θ scans
Absorption correction: ψ scan (North et al., 1968)
Tmin = 0.962, Tmax = 0.972
1686 measured reflections
888 independent reflections 805 reflections with I > 2σ(I)
Rint = 0.032
θmax = 26.0°, θmin = 3.0°
h = −10→0
k = −10→0
l = −12→12
3 standard reflections every 200 reflections intensity decay: none
Refinement
Refinement on F2
Least-squares matrix: full
R[F2 > 2σ(F2)] = 0.034
wR(F2) = 0.082
S = 1.08 888 reflections 92 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H-atom parameters constrained
w = 1/[σ2(F
o2) + (0.04P)2 + 0.05P]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max < 0.001
Δρmax = 0.18 e Å−3
Δρmin = −0.13 e Å−3
Extinction correction: SHELXL97, Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4
Special details
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2,
conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used
only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2
are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
O1 0.09773 (18) 0.63916 (19) 0.91853 (16) 0.0617 (5) O2 0.15140 (17) 0.96009 (17) 0.78132 (14) 0.0522 (4) O3 0.13615 (18) 0.57691 (18) 0.71336 (14) 0.0569 (5)
H3 0.0454 0.5425 0.7197 0.085*
N1 0.34804 (18) 0.85087 (17) 0.89286 (15) 0.0403 (4)
H1 0.4156 0.8568 0.9553 0.048*
C1 0.2072 (3) 1.2358 (2) 0.9254 (3) 0.0691 (7)
H1A 0.1462 1.2468 0.8509 0.083*
H1B 0.2225 1.3186 0.9801 0.083*
C2 0.2716 (3) 1.1042 (2) 0.9532 (2) 0.0504 (5)
H2 0.3322 1.0955 1.0282 0.060*
C3 0.2516 (2) 0.9678 (2) 0.86977 (17) 0.0396 (5) C4 0.3424 (2) 0.7135 (2) 0.81552 (18) 0.0393 (5)
H4 0.3669 0.7393 0.7255 0.047*
C5 0.4649 (3) 0.5992 (3) 0.8644 (3) 0.0584 (6)
H5A 0.5697 0.6435 0.8601 0.088*
H5B 0.4616 0.5092 0.8116 0.088*
H5C 0.4409 0.5726 0.9522 0.088*
C6 0.1766 (2) 0.6428 (2) 0.82196 (18) 0.0378 (4)
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
supporting information
sup-3 Acta Cryst. (2007). E63, o191–o192
Geometric parameters (Å, º)
N1—C3 1.327 (2) O2—C3 1.243 (2)
N1—C4 1.447 (2) O3—C6 1.309 (2)
N1—H1 0.8600 O3—H3 0.8200
C1—C2 1.307 (3) C4—C6 1.521 (2)
C1—H1A 0.9300 C4—C5 1.521 (3)
C1—H1B 0.9300 C4—H4 0.9800
O1—C6 1.198 (2) C5—H5A 0.9600
C2—C3 1.485 (3) C5—H5B 0.9600
C2—H2 0.9300 C5—H5C 0.9600
C3—N1—C4 121.60 (15) N1—C4—C5 110.07 (16)
C3—N1—H1 119.2 C6—C4—C5 109.35 (16)
C4—N1—H1 119.2 N1—C4—H4 109.1
C2—C1—H1A 120.0 C6—C4—H4 109.1
C2—C1—H1B 120.0 C5—C4—H4 109.1
H1A—C1—H1B 120.0 C4—C5—H5A 109.5
C1—C2—C3 122.5 (2) C4—C5—H5B 109.5
C1—C2—H2 118.7 H5A—C5—H5B 109.5
C3—C2—H2 118.7 C4—C5—H5C 109.5
C6—O3—H3 109.5 H5A—C5—H5C 109.5
O2—C3—N1 120.06 (18) H5B—C5—H5C 109.5
O2—C3—C2 123.21 (17) O1—C6—O3 124.20 (17)
N1—C3—C2 116.73 (17) O1—C6—C4 123.36 (18)
N1—C4—C6 110.20 (15) O3—C6—C4 112.24 (16)
C4—N1—C3—O2 1.2 (3) C3—N1—C4—C5 −179.64 (18) C4—N1—C3—C2 −178.35 (17) N1—C4—C6—O1 −38.8 (3) C1—C2—C3—O2 −13.2 (4) C5—C4—C6—O1 82.3 (2) C1—C2—C3—N1 166.4 (2) N1—C4—C6—O3 146.07 (16) C3—N1—C4—C6 −59.0 (2) C5—C4—C6—O3 −92.8 (2)
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
N1—H1···O1i 0.86 2.01 2.861 (2) 171
O3—H3···O2ii 0.82 1.80 2.616 (2) 175