organic papers
o342
van Blerk and Kruger C4H14N22+2Br doi:10.1107/S1600536806053736 Acta Cryst.(2007). E63, o342–o344 Acta Crystallographica Section E
Structure Reports Online
ISSN 1600-5368
Butane-1,4-diammonium dibromide
Charmaine van Blerk* and Gert J. Kruger
Department of Chemistry, University of nesburg, PO Box 524, Auckland Park, Johan-nesburg 2006, South Africa
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study
T= 293 K
Mean(C–C) = 0.006 A˚
Rfactor = 0.045
wRfactor = 0.131
Data-to-parameter ratio = 28.5
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
Received 1 December 2006 Accepted 11 December 2006
#2007 International Union of Crystallography All rights reserved
The crystal structure of butane-1,4-diammonium dibromide, C4H14N2
2+
2Br, exhibits ionic–inorganic layers separated by organic hydrocarbon layers. The organic hydrocarbon layers pack in a stacked herring-bone manner with hydrogen bonds to the bromide ions. The cation sits on a centre of inversion. The structure is compared with that of butane-1,4-diammo-nium dichloride and butane-1,4-diammobutane-1,4-diammo-nium diiodide.
Comment
As part of an ongoing study of the structural characteristics of organic–inorganic layered diammonium salts, the crystal structure of butane-1,4-diammonium dibromide, (I), was determined. A search of the Cambridge Structural Database (Version 5.27, November 2005 release; Allen, 2002) revealed that the crystal structure of butane-1,4-diammonium dichloride was redetermined and published 26 years ago (Chandrasekhar & Pattabhi, 1980). Recently, the crystal structure of butane-1,4-diammonium diiodide was studied and published (Lemmerer & Billing, 2006). The present study completes the series of halide salts and shows all three compounds to be isostructural. The dichloride structure was shown to exhibit a short N Cl non-bonded contact that links the two-dimensional layers along theadirection. Butane-1,4-diammonium dibromide exhibits the same interaction.
[image:1.610.239.421.469.518.2] [image:1.610.229.433.597.710.2]The butane-1,4-diammonium cation sits on a centre of inversion and therefore the asymmetric unit contains one anion and half of the cation (Fig. 1).
Figure 1
Fig. 2 illustrates the packing of the compound viewed down the c axis. The packing arrangement shown is one-dimen-sional, with a single layer of butane-1,4-diammonium cations sandwiched between two layers of bromide anions, thus forming an alternating organic–inorganic structure. The butane-1,4-diammonium cations pack in a herring-bone manner (Fig. 3).
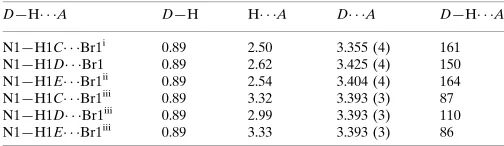
Fig. 4 clearly shows the occurrence of four short N Br non-bonded contacts. Three of these contacts may be ascribed to charge-assisted hydrogen-bond formation and the fourth is a short contact directed approximately along the extension of the C—N bond. This fourth contact may be viewed as also involving H atoms since the Branion is at similar distances from the three atoms H1C, H1Dand H1E(Table 1).
The N—H Br hydrogen bonds (Table 1) form an exten-sive network. Two ammonium H atoms, H1Cand H1D, form hydrogen-bonded rings with graph-set notationR22(8) while
hydrogen-bonded rings with graph-set notation R23(13)
involve all three ammonium H atoms. The smaller ring is rectangular in shape, with both the short and the long edges of the rectangle sharing sides with the larger ring. The larger ring has five sides, of which one is the butane-1,4-diammonium cation. All the edges of the rings are shared, forming an infi-nite network in theacplane.
Experimental
The title compound was prepared by adding 1,4-diaminobutane (0.050 g, 0.567 mmol) to 47% hydrobromic acid (2 ml, 37 mmol) in a sample vial. The mixture was then refluxed at 363 K for 2 h. The solution was cooled slowly at 2 K h1to room temperature.
Colour-less crystals of butane-1,4-diammonium dibromide were collected.
Crystal data
C4H14N22+2Br Mr= 249.99 Monoclinic,P21=c
a= 4.7107 (9) A˚
b= 8.5491 (15) A˚
c= 11.229 (2) A˚
= 93.037 (4) V= 451.57 (14) A˚3
Z= 2
Dx= 1.839 Mg m
3
MoKradiation
= 8.90 mm1
T= 293 (2) K Block, colourless 0.440.300.28 mm
Data collection
Bruker SMART CCD area-detector diffractometer
’and!scans
Absorption correction: multi-scan (SADABS; Sheldrick, 2004)
Tmin= 0.056,Tmax= 0.190
(expected range = 0.024–0.083)
2321 measured reflections 1113 independent reflections 942 reflections withI> 2(I)
Rint= 0.024
max= 28.3
Refinement
Refinement onF2 R[F2> 2(F2)] = 0.045
wR(F2) = 0.131
S= 0.99 1113 reflections 39 parameters
H-atom parameters constrained
w= 1/[2
(Fo2) + (0.0984P)2]
whereP= (Fo2+ 2Fc2)/3
(/)max< 0.001
max= 1.50 e A˚ 3
min=1.64 e A˚ 3
Extinction correction:SHELXL97
Extinction coefficient: 0.45 (3)
organic papers
Acta Cryst.(2007). E63, o342–o344 van Blerk and Kruger C
4H14N22+2Br
o343
Figure 2
Packing diagram of (I), viewed down thecaxis. Each layer of butane-1,4-diammonium cations is sandwiched by layers of bromide ions.
Figure 3
[image:2.610.46.294.539.697.2]The two-dimensional hydrogen-bonded (dotted lines) network in (I), viewed down theaaxis.
Figure 4
A view of (I), showing the fourth short contact with particular focus on the hydrogen bonding of the three ammonium H atoms with the Br
Table 1
Hydrogen-bond geometry (A˚ ,).
D—H A D—H H A D A D—H A
N1—H1C Br1i
0.89 2.50 3.355 (4) 161
N1—H1D Br1 0.89 2.62 3.425 (4) 150
N1—H1E Br1ii
0.89 2.54 3.404 (4) 164
N1—H1C Br1iii
0.89 3.32 3.393 (3) 87
N1—H1D Br1iii
0.89 2.99 3.393 (3) 110
N1—H1E Br1iii 0.89 3.33 3.393 (3) 86
Symmetry codes: (i)xþ1;y;zþ1; (ii)x;yþ1 2;zþ
1
2; (iii)x;y;zþ1.
H atoms were geometrically positioned and refined in the riding-model approximation, with C—H = 0.97 A˚ , N—H = 0.89 A˚, and
Uiso(H) = 1.2Ueq(C) or 1.5Ueq(N). The highest peak in the final
difference map is 0.82 A˚ from Br1 and the deepest hole is 0.90 A˚ from Br1.
Data collection:SMART(Bruker, 1998); cell refinement: SAINT-Plus(Bruker, 1999); data reduction:SAINT-Plus; program(s) used to solve structure: SHELXS97 (Sheldrick, 1997); program(s) used to refine structure:SHELXL97(Sheldrick, 1997); molecular graphics:
ORTEP-3 for Windows(Farrugia, 1997) andMercury(Macraeet al., 2006); software used to prepare material for publication: WinGX
(Farrugia, 1999), PLATON (Spek, 2003) and pubICIF (Westrip, 2006).
The authors acknowledge the University of the Witwa-tersrand for their facilities and the use of the diffractometer in the Jan Boeyens Structural Chemistry Laboratory.
References
Allen, F. H. (2002).Acta Cryst.B58, 380–388.
Bruker (1998). SMART-NT. Version 5.050. Bruker AXS Inc., Madison, Wisconsin, USA.
Bruker (1999). SAINT-Plus. Version 6.02. Bruker AXS Inc., Madison, Wisconsin, USA.
Chandrasekhar, K. & Pattabhi, V. (1980).Acta Cryst.B36, 2486–2488. Farrugia, L. J. (1997).J. Appl. Cryst.30, 565.
Farrugia, L. J. (1999).J. Appl. Cryst.32, 837–838.
Lemmerer, A. & Billing, D. G. (2006).Acta Cryst.E62, o1954–o1956. Macrae, C. F., Edgington, P. R., McCabe, P., Pidcock, E., Shields, G. P., Taylor,
R., Towler, M. &van de Streek, J. (2006).J. Appl. Cryst.39, 453–457. Sheldrick, G. M. (1997). SHELXS97 and SHELXL97. University of
Go¨ttingen, Germany.
Sheldrick, G. M. (2004).SADABS. Version 2004/1. University of Go¨ttingen, Germany.
Spek, A. L. (2003).J. Appl. Cryst.36, 7–13. Westrip, S. P. (2006).publCIF. In preparation.
organic papers
o344
van Blerk and Kruger Csupporting information
sup-1
Acta Cryst. (2007). E63, o342–o344
supporting information
Acta Cryst. (2007). E63, o342–o344 [https://doi.org/10.1107/S1600536806053736]
Butane-1,4-diammonium dibromide
Charmaine van Blerk and Gert J. Kruger
Butane-1,4-diammonium dibromide
Crystal data
C4H14N22+·2Br− Mr = 249.99
Monoclinic, P21/c
Hall symbol: -P 2ybc
a = 4.7107 (9) Å
b = 8.5491 (15) Å
c = 11.229 (2) Å
β = 93.037 (4)°
V = 451.57 (14) Å3
Z = 2
F(000) = 244
Dx = 1.839 Mg m−3
Mo Kα radiation, λ = 0.71073 Å
Cell parameters from 669 reflections
θ = 3.0–28.3°
µ = 8.90 mm−1
T = 293 K
Block, colourless 0.44 × 0.30 × 0.28 mm
Data collection
Bruker SMART CCD area-detector diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
φ and ω scans
Absorption correction: multi-scan (SADABS; Sheldrick, 2004) Tmin = 0.056, Tmax = 0.190
2321 measured reflections 1113 independent reflections 942 reflections with I > 2σ(I) Rint = 0.024
θmax = 28.3°, θmin = 3.0°
h = −6→6
k = −6→11
l = −11→14
Refinement
Refinement on F2
Least-squares matrix: full R[F2 > 2σ(F2)] = 0.045 wR(F2) = 0.131
S = 0.99
1113 reflections 39 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H-atom parameters constrained w = 1/[σ2(F
o2) + (0.0984P)2] where P = (Fo2 + 2Fc2)/3 (Δ/σ)max < 0.001
Δρmax = 1.50 e Å−3 Δρmin = −1.64 e Å−3
supporting information
sup-2
Acta Cryst. (2007). E63, o342–o344
Special details
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used
only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2
are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
C1 0.5747 (8) 0.3000 (4) 0.5718 (4) 0.0324 (8)
H1A 0.6773 0.2556 0.5073 0.039*
H1B 0.7129 0.3337 0.6337 0.039*
C2 0.4044 (9) 0.4393 (5) 0.5263 (4) 0.0331 (9)
H2A 0.3059 0.4855 0.5913 0.040*
H2B 0.2630 0.4053 0.4659 0.040*
N1 0.3869 (8) 0.1784 (4) 0.6208 (4) 0.0366 (8)
H1C 0.4915 0.0970 0.6459 0.055*
H1D 0.2600 0.1475 0.5641 0.055*
H1E 0.2971 0.2181 0.6817 0.055*
Br1 0.11369 (9) 0.09905 (4) 0.33855 (4) 0.0370 (3)
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
C1 0.0310 (18) 0.0311 (18) 0.035 (2) 0.0005 (15) 0.0007 (15) 0.0005 (16)
C2 0.035 (2) 0.0295 (17) 0.035 (2) 0.0013 (16) 0.0034 (16) 0.0034 (16)
N1 0.0414 (19) 0.0280 (16) 0.0407 (19) −0.0001 (14) 0.0041 (14) 0.0020 (14)
Br1 0.0416 (4) 0.0366 (4) 0.0327 (4) −0.00146 (15) 0.0021 (2) 0.00391 (14)
Geometric parameters (Å, º)
C1—N1 1.489 (5) C2—H2A 0.970
C1—C2 1.510 (5) C2—H2B 0.970
C1—H1A 0.970 N1—H1C 0.890
C1—H1B 0.970 N1—H1D 0.890
C2—C2i 1.514 (8) N1—H1E 0.890
N1—C1—C2 111.1 (3) C1—C2—H2B 109.5
N1—C1—H1A 109.4 C2i—C2—H2B 109.5
C2—C1—H1A 109.4 H2A—C2—H2B 108.1
N1—C1—H1B 109.4 C1—N1—H1C 109.5
C2—C1—H1B 109.4 C1—N1—H1D 109.5
H1A—C1—H1B 108.0 H1C—N1—H1D 109.5
C1—C2—C2i 110.9 (4) C1—N1—H1E 109.5
supporting information
sup-3
Acta Cryst. (2007). E63, o342–o344
C2i—C2—H2A 109.5 H1D—N1—H1E 109.5
N1—C1—C2—C2i −178.5 (4)
Symmetry code: (i) −x+1, −y+1, −z+1.
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
N1—H1C···Br1ii 0.89 2.50 3.355 (4) 161
N1—H1D···Br1 0.89 2.62 3.425 (4) 150
N1—H1E···Br1iii 0.89 2.54 3.404 (4) 164
N1—H1C···Br1iv 0.89 3.32 3.393 (3) 87
N1—H1D···Br1iv 0.89 2.99 3.393 (3) 110
N1—H1E···Br1iv 0.89 3.33 3.393 (3) 86