organic papers
o1088
Graham Smithet al. C16H24N3O5 DOI: 10.1107/S160053680201601X Acta Cryst.(2002). E58, o1088±o1090 Acta Crystallographica Section EStructure Reports
Online
ISSN 1600-5368
Ethylenediammonium 4-nitroanthranilate
dihydrate
Graham Smith,a* Urs D. Wermuthaand Jonathan M. Whiteb
aCentre for Instrumental and Developmental
Chemistry, Queensland University of Technology, GPO Box 2434, Brisbane 4001, Australia, andbSchool of Chemistry, University of Melbourne, Parkville, 3052, Australia
Correspondence e-mail: g.smith@qut.edu.au
Key indicators Single-crystal X-ray study
T= 293 K
Mean(C±C) = 0.002 AÊ
Rfactor = 0.045
wRfactor = 0.137
Data-to-parameter ratio = 14.2
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2002 International Union of Crystallography Printed in Great Britain ± all rights reserved
The crystal structure of ethylenediammonium 4-nitro-anthranilate dihydrate, [(C2H10N2)2+2(C7H5N2O4)ÿ2(H2O)],
shows a three-dimensional hydrogen-bonded polymer in which both of the amine groups of ethylenediamine are protonated and each gives a total of four hydrogen-bonded interactions with oxygen and amine N atoms of the anthranilate anions as well as with the water molecules. The centrosymmetrically related anthranilate species are also linked directly to the water molecules, and give a double-chain structure down thebaxis.
Comment
Ethylenediamine (ethane-1,2-diamine = en) reacts with acids to give stable crystalline salts, and because of the relative similarity of the dissociation constants of en (pKa1= 7.3; pKa2
= 10.1), both amine groups are protonated, even in reactions with weak organic acids. With the enH22+species, as is the case
with protonated primary amine groups, the ÐNH3+hydrogens
may be involved in up to six intermolecular hydrogen-bonding interactions with suitable acceptor atoms. The resulting hydrogen-bonded polymer structures acquire considerable crystal stability together with enhanced melting points. This is seen with the en salts of the relatively strong nitro-substituted benzoic acids,e.g.3,5-dinitrobenzoic acid (DNBA), [(enH2)2+
2(DNBA)ÿ] (Nethaji et al., 1992; Lynch et al., 1994), while
crystallization often includes lattice water molecules, increasing the structure-making e.g. ethylenediammonium 5-nitrosalicylate hydrate, [(enH2)2+ 2(5-NSA)ÿH2O] (Smith
& Hartono, 2002). With the bifunctional 3,5-dinitrosalicylic acid (DNSA), the rare occurrence of the (DNSA)2ÿ species
has been observed in the salt [(enH2)2+(DNSA)2ÿ] (Smithet
al., 2002). Our interest lies in the characterization of the hydrogen-bonding interactions of the nitro-substituted aromatic acids with Lewis bases, The structure of the title compound, obtained from the reaction of 4-nitroanthranilic acid (4-NAA) with en as a hydrate, described in terms of the centrosymmetric molecular unit (the unit cell contents) [(en)2+
2(4-NAA)ÿ2(H
2O)], (I), is reported here.
The structure determination of (I) shows that both the primary amine groups of ethylenediamine are protonated (Fig. 1). A hydrogen bond is found between an amine±H and an oxygen of the carboxyl group of the 4-NAA anion [N1ÐH1A O2; 2.694 (2) AÊ]. The 4-NAA anions, one unit cell apart along thebaxis, are linked by N1Ð H1B O1(x, 1 +y, z) hydrogen bonds to form an in®nite one-dimensional chain. The molecules in the chain are linked to those in the inversion-related chain (1ÿx,ÿy, 1ÿz) by the water molecules, through OÐH O hydrogen bonds, to form a double-chain structure (Fig. 2). These 4-NAA anion chains stack down theaaxis and are linked by the (enH2)2+cations as
well as the water molecules, giving a three-dimensional polymer (Fig. 3). A full hydrogen-bond listing is given in Table 1.
Experimental
The synthesis of the title compound was carried out by heating, under re¯ux for 10 min, 1 mmol quantities of 4-nitroanthranilic acid (2-amino-4-nitrobenzoic acid = 4-NAA) and ethylenediamine (en) in 50 ml of 80% ethanol/water. After concentration toca. 30 ml, partial room temperature evaporation of the hot-®ltered solution gave red crystals.
Crystal data
C2H10N22+2C7H5N2O4ÿ2H2O
Mr= 460.42 Triclinic, P1
a= 6.6473 (5) AÊ
b= 7.0748 (5) AÊ
c= 11.2317 (8) AÊ
= 76.686 (2)
= 77.660 (2)
= 89.525 (2)
V= 501.67 (6) AÊ3
Z= 1
Dx= 1.524 Mg mÿ3 MoKradiation Cell parameters from 2947
re¯ections
= 3.0±28.3
= 0.13 mmÿ1
T= 293 (2) K Plate, red
0.400.300.20 mm
Data collection
Bruker SMART CCD area-detector diffractometer
'and!scans
Absorption correction: none 4462 measured re¯ections 2475 independent re¯ections
2052 re¯ections withI>2(I)
Rint= 0.046 max= 28.6
h=ÿ8!8
k=ÿ9!9
l=ÿ15!14
Re®nement
Re®nement onF2
R[F2> 2(F2)] = 0.045
wR(F2) = 0.137
S= 1.07 2475 re¯ections 174 parameters
H atoms treated by a mixture of independent and constrained re®nement
w= 1/[2(F
o2) + (0.0745P)2 + 0.0989P]
whereP= (Fo2+ 2Fc2)/3 (/)max= 0.010
max= 0.26 e AÊÿ3
min=ÿ0.23 e AÊÿ3
Table 1
Hydrogen-bonding geometry (AÊ,).
DÐH A DÐH H A D A DÐH A
N1ÐH1A O2 0.88 (2) 2.04 (2) 2.6935 (17) 130.8 (18) N1ÐH1B O1i 0.82 (2) 2.12 (2) 2.9258 (17) 167.8 (17)
N3ÐH3A O5ii 0.90 (2) 1.95 (2) 2.7960 (18) 157.5 (19)
N3ÐH3B O3iii 0.87 (2) 2.41 (2) 3.0564 (18) 130.9 (17)
N3ÐH3B N1iv 0.87 (2) 2.52 (2) 3.1847 (18) 133.8 (18)
N3ÐH3C O2i 0.917 (19) 1.937 (19) 2.8250 (17) 162.5 (14)
O5ÐH5A O1i 0.84 (2) 1.91 (2) 2.7429 (16) 179 (2)
O5ÐH5B O2v 0.79 (3) 2.18 (3) 2.9647 (16) 171 (3)
C6ÐH6 O1 0.93 2.41 2.7454 (16) 101
Symmetry codes: (i) x;1y;z; (ii) ÿx;1ÿy;1ÿz; (iii) xÿ1;y;1z; (iv) 1ÿx;1ÿy;1ÿz; (v) 1ÿx;ÿy;1ÿz.
Acta Cryst.(2002). E58, o1088±o1090 Graham Smithet al. C16H24N3O5
o1089
organic papers
Figure 1
Molecular con®guration and atom-naming scheme for the individual 4-NAA anion, the en cation and the water species in (I). Non-hydrogen
atoms are shown as 40% probability ellipsoids Figure 2A view of the double-chain formation by 4-NAA and water molecules.
Figure 3
organic papers
o1090
Graham Smithet al. C16H24N3O5 Acta Cryst.(2002). E58, o1088±o1090H atoms involved in hydrogen-bonding interactions (H1A, H1B, H3A, H3B, H3C, H5A, H5B) were located from a difference Fourier map and their positional and isotropic displacement parameters were re®ned. Others were included in the re®nement at calculated posi-tions as riding models. For re®ned H atoms, the NÐH range is 0.87 (2)±0.92 (2) AÊ; the OÐH (water) values are 0.79 (3) and 0.84 (2) AÊ.
Data collection:SMART(Bruker, 2000); cell re®nement:SMART; data reduction: SAINT (Bruker, 1999); program(s) used to solve structure: SHELXTL (Bruker, 1997); program(s) used to re®ne structure:SHELXTL; molecular graphics:PLATON(Spek, 1999); software used to prepare material for publication:SHELXTL.
The authors acknowledge ®nancial support from The Centre for Instrumental and Developmental Chemistry
(Queensland University of Technology) and The University of Melbourne.
References
Bruker (1997). SHELXTL Version 5.10. Bruker AXS Inc., Madison, Wisconsin, USA.
Bruker (1999).SAINT. Version 6.02. Bruker AXS Inc., Madison, Wisconsin, USA.
Bruker (2000).SMART. Version 5.55. Bruker AXS Inc., Madison, Wisconsin, USA.
Lynch, D. E, Smith, G., Byriel, K. A. & Kennard, C. H. L. (1994).Acta Cryst.
C50, 1291±1294.
Nethaji, M., Pattabhi V., Chhabra, N & Poonia, N. S. (1992).Acta Cryst.C48, 2207±2209.
Smith, G., Wermuth, U. D., Bott, R. C., Healy, P. C. & White, J. M. (2002).Aust. J. Chem.55, 349±356.
Smith, G. & Hartono, A. (2002). Unpublished results.
supporting information
sup-1
Acta Cryst. (2002). E58, o1088–o1090supporting information
Acta Cryst. (2002). E58, o1088–o1090 [doi:10.1107/S160053680201601X]
Ethylenediammonium 4-nitroanthranilate dihydrate
Graham Smith, Urs D. Wermuth and Jonathan M. White
S1. Comment
Ethylenediamine (ethane-1,2-diamine = EN) reacts with acids to give stable crystalline salts and because of the relative
closeness of the dissociation constants of EN (pKa1 = 7.3; pKa2 = 10.1), both amine groups are protonated even in
reactions with weak organic acids. With the EN2+ species, as is the case with protonated primary amine groups, the —
NH3+ protons may be involved in up to six intermolecular hydrogen-bonding interactions with suitable acceptor atoms.
The resulting hydrogen-bonded polymer structures acquire considerable crystal stability together with enhanced melting
points. This is seen with the EN salts of the relatively strong nitro-substituted benzoic acids, e.g. 3,5-dinitrobenzoic acid
(DNBA), [(EN)2+ 2(DNBA)−] (Nethaji et al., 1992; Lynch et al., 1994), while crystallization often includes lattice water
molecules, increasing the structure-making e.g. ethylenediammonium 5-nitrosalicylate hydrate, [(EN)2+ 2(5-NSA)+. H 2O]
(Smith & Hartono, 2002). With the bifunctional 3,5-dinitrosalicylic acid (DNSA), the rare occurrence of the (DNSA)2−
species has been observed in the salt [(EN)2+ (DNSA)2−] (Smith et al., 2002). Our interest lies in the characterization of
the hydrogen-bonding interactions of the nitro-substituted aromatic acids with Lewis bases and the structure of the title
compound from the reaction of 4-nitroanthranilic acid (4-NAA) with EN, the hydrate, best described in terms of the
centrosymmetric molcular unit (the unit-cell contents) [(EN)2+ 2(4-NAA)−·2(H
2O)], (I), is reported here.
The structure determination of (I) shows that both the primary amine groups of ethylenediamine are protonated (Fig. 1).
An intramolecular hydrogen bond is found between an amine-H and an oxygen of the carboxyl group of the 4-NAA anion
[N1—H1A···O2; 2.694 (2) Å]. The 4-NAA anions translated one unit along the b cell direction are linked by N1—
H1B···O1(x, 1 + y, z) hydrogen bonds to form an infinite one dimensional chain. The molecules in the chain are linked to
those in the inversion related chain (1 − x, −y, 1 − z) by the water molecules through O—H···O hydrogen bonds to form a
double chain structure (Fig. 2). These 4-NAA anion chains stack down the a cell direction and are linked by the EN
cations as well as the water molecules giving a three-dimensional polymer (Fig. 3). A full hydrogen bond listing is given
in Table 1.
S2. Experimental
The synthesis of the title compound was carried out by heating under reflux for 10 min, 1 mmol quantities of
4-nitro-anthranilic acid (2-amino-4-nitrobenzoic acid = 4-NAA) and ethylenediamine (EN) in 50 ml of 80% ethanol/water. After
concentration to ca 30 ml, partial room temperature evaporation of the hot-filtered solution gave red crystals.
S3. Refinement
Hydrogen atoms involved in hydrogen-bonding interactions (H1A, H1B, H3A, H3B, H3C, H5A, H5B) were located from
a difference Fourier map and their positional and isotropic thermal parameters were refined. Others were included in the
refinement at calculated positions as riding models. For refined hydrogen atoms, the N–H range is 0.87 (2)–0.92 (2) Å;
supporting information
[image:5.610.130.486.70.282.2]sup-2
Acta Cryst. (2002). E58, o1088–o1090Figure 1
Molecular configuration and atom-naming scheme for the individual 4-NAA anion, the EN cation and the water species
in (I). Atoms are shown as 40% probability ellipsoids
Figure 2
[image:5.610.132.483.339.644.2]supporting information
[image:6.610.132.474.70.314.2]sup-3
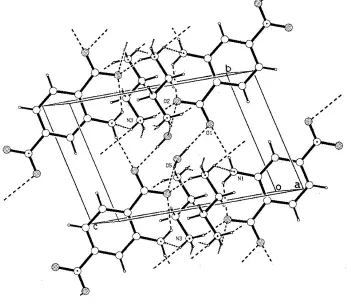
Acta Cryst. (2002). E58, o1088–o1090Figure 3
Packing in the unit cell, viewed down b, showing hydrogen-bonding associations as broken lines.
(I)
Crystal data
C2H10N22+·2C7H5N2O4−·2H2O
Mr = 460.42 Triclinic, P1 Hall symbol: -P 1 a = 6.6473 (5) Å b = 7.0748 (5) Å c = 11.2317 (8) Å α = 76.686 (2)° β = 77.660 (2)° γ = 89.525 (2)° V = 501.67 (6) Å3
Z = 1 F(000) = 242 Dx = 1.524 Mg m−3
Melting point = 493.6–495.4 K Mo Kα radiation, λ = 0.71073 Å Cell parameters from 2947 reflections θ = 3.0–28.3°
µ = 0.13 mm−1
T = 293 K Plate, red
0.40 × 0.30 × 0.20 mm
Data collection
Bruker SMART CCD area detector diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
φ and ω scans
4462 measured reflections 2475 independent reflections
2052 reflections with I > 2σ(I) Rint = 0.046
θmax = 28.6°, θmin = 1.9°
h = −8→8 k = −9→9 l = −15→14
Refinement
Refinement on F2
Least-squares matrix: full R[F2 > 2σ(F2)] = 0.045
wR(F2) = 0.137
S = 1.07
2475 reflections 174 parameters 0 restraints
supporting information
sup-4
Acta Cryst. (2002). E58, o1088–o1090Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H atoms treated by a mixture of independent and constrained refinement
w = 1/[σ2(F
o2) + (0.0745P)2 + 0.0989P]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max = 0.010
Δρmax = 0.26 e Å−3
Δρmin = −0.23 e Å−3
Special details
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2,
conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used
only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2
are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
C1 0.64571 (17) −0.04597 (16) 0.15627 (10) 0.0277 (3)
C2 0.66670 (18) 0.15338 (17) 0.15449 (11) 0.0301 (3)
C3 0.7519 (2) 0.28286 (17) 0.03922 (12) 0.0344 (3)
H3 0.7677 0.4149 0.0351 0.041*
C4 0.81153 (19) 0.21337 (19) −0.06693 (11) 0.0336 (3)
C5 0.79504 (19) 0.0192 (2) −0.06782 (11) 0.0345 (3)
H5 0.8380 −0.0239 −0.1410 0.041*
C6 0.71171 (18) −0.10772 (17) 0.04521 (11) 0.0312 (3)
H6 0.6990 −0.2394 0.0475 0.037*
C7 0.54568 (19) −0.19649 (17) 0.27287 (11) 0.0325 (3)
C8 0.0253 (2) 0.94508 (18) 0.56105 (11) 0.0372 (3)
H8A 0.1297 1.0184 0.5823 0.045*
H8B −0.0970 0.9292 0.6280 0.045*
N1 0.6138 (2) 0.22588 (18) 0.25992 (11) 0.0425 (3)
N2 0.90109 (19) 0.35319 (19) −0.18554 (10) 0.0447 (3)
N3 0.1014 (2) 0.75257 (16) 0.54837 (11) 0.0375 (3)
H3A 0.002 (3) 0.682 (3) 0.5334 (17) 0.053 (5)*
H3B 0.133 (3) 0.695 (3) 0.619 (2) 0.062 (5)*
H3C 0.219 (3) 0.763 (2) 0.4869 (17) 0.047 (4)*
O1 0.5325 (2) −0.36820 (14) 0.26359 (10) 0.0555 (3)
O2 0.47940 (18) −0.14314 (15) 0.37310 (9) 0.0472 (3)
O3 0.9185 (3) 0.52434 (19) −0.18427 (12) 0.0736 (5)
O4 0.9547 (2) 0.2929 (2) −0.28036 (10) 0.0613 (4)
O5 0.27641 (17) 0.39858 (16) 0.46802 (11) 0.0466 (3)
H5A 0.355 (3) 0.470 (3) 0.406 (2) 0.058 (5)*
H1A 0.531 (3) 0.151 (3) 0.3245 (19) 0.054 (5)*
H1B 0.597 (3) 0.343 (3) 0.2492 (17) 0.049 (5)*
supporting information
sup-5
Acta Cryst. (2002). E58, o1088–o1090Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
C1 0.0282 (5) 0.0256 (6) 0.0268 (6) 0.0010 (4) −0.0046 (4) −0.0023 (4)
C2 0.0330 (6) 0.0280 (6) 0.0276 (6) 0.0008 (4) −0.0048 (4) −0.0045 (4)
C3 0.0425 (7) 0.0254 (6) 0.0325 (6) −0.0030 (5) −0.0079 (5) −0.0012 (4)
C4 0.0323 (6) 0.0383 (7) 0.0252 (6) −0.0042 (5) −0.0058 (4) 0.0019 (5)
C5 0.0340 (6) 0.0434 (7) 0.0264 (6) 0.0014 (5) −0.0052 (4) −0.0103 (5)
C6 0.0329 (6) 0.0282 (6) 0.0327 (6) 0.0018 (4) −0.0067 (5) −0.0080 (4)
C7 0.0329 (6) 0.0289 (6) 0.0311 (6) −0.0020 (4) −0.0039 (5) −0.0008 (4)
C8 0.0486 (7) 0.0330 (6) 0.0293 (6) 0.0010 (5) −0.0055 (5) −0.0084 (5)
N1 0.0616 (8) 0.0286 (6) 0.0329 (6) −0.0001 (5) 0.0005 (5) −0.0084 (4)
N2 0.0452 (7) 0.0520 (7) 0.0299 (6) −0.0107 (5) −0.0082 (5) 0.0053 (5)
N3 0.0455 (6) 0.0309 (6) 0.0310 (6) −0.0003 (5) −0.0021 (5) −0.0027 (4)
O1 0.0759 (8) 0.0254 (5) 0.0504 (6) −0.0053 (5) 0.0106 (5) −0.0016 (4)
O2 0.0616 (7) 0.0402 (5) 0.0307 (5) −0.0120 (5) 0.0066 (4) −0.0049 (4)
O3 0.1121 (12) 0.0524 (7) 0.0433 (7) −0.0338 (7) −0.0106 (7) 0.0104 (5)
O4 0.0693 (8) 0.0758 (9) 0.0272 (5) 0.0007 (6) 0.0018 (5) 0.0001 (5)
O5 0.0448 (6) 0.0462 (6) 0.0394 (6) −0.0088 (5) −0.0045 (5) 0.0048 (5)
Geometric parameters (Å, º)
C1—C6 1.3975 (16) C8—N3 1.4758 (17)
C1—C2 1.4135 (16) C8—C8i 1.517 (2)
C1—C7 1.5128 (15) C8—H8A 0.97
C2—N1 1.3740 (16) C8—H8B 0.97
C2—C3 1.4089 (17) N1—H1A 0.88 (2)
C3—C4 1.3730 (18) N1—H1B 0.82 (2)
C3—H3 0.93 N2—O4 1.2195 (17)
C4—C5 1.3812 (19) N2—O3 1.2210 (18)
C4—N2 1.4736 (15) N3—H3A 0.90 (2)
C5—C6 1.3812 (18) N3—H3B 0.87 (2)
C5—H5 0.93 N3—H3C 0.915 (18)
C6—H6 0.93 O5—H5A 0.84 (2)
C7—O1 1.2483 (16) O5—H5B 0.79 (3)
C7—O2 1.2588 (16)
C6—C1—C2 119.33 (10) O2—C7—C1 118.94 (11)
C6—C1—C7 117.84 (10) N3—C8—C8i 109.84 (13)
C2—C1—C7 122.78 (11) N3—C8—H8A 109.7
N1—C2—C3 118.54 (11) C8i—C8—H8A 109.7
N1—C2—C1 123.30 (11) N3—C8—H8B 109.7
C3—C2—C1 118.13 (11) C8i—C8—H8B 109.7
C4—C3—C2 119.73 (11) H8A—C8—H8B 108.2
C4—C3—H3 120.1 C2—N1—H1A 115.8 (13)
C2—C3—H3 120.1 C2—N1—H1B 117.0 (12)
C3—C4—C5 123.44 (11) H1A—N1—H1B 116.3 (18)
supporting information
sup-6
Acta Cryst. (2002). E58, o1088–o1090C5—C4—N2 118.43 (12) O4—N2—C4 118.63 (13)
C4—C5—C6 116.82 (11) O3—N2—C4 118.38 (12)
C4—C5—H5 121.6 C8—N3—H3A 109.5 (12)
C6—C5—H5 121.6 C8—N3—H3B 107.6 (13)
C5—C6—C1 122.54 (11) H3A—N3—H3B 109.6 (18)
C5—C6—H6 118.7 C8—N3—H3C 111.5 (11)
C1—C6—H6 118.7 H3A—N3—H3C 111.2 (15)
O1—C7—O2 123.55 (11) H3B—N3—H3C 107.3 (18)
O1—C7—C1 117.51 (11) H5A—O5—H5B 107 (2)
C6—C1—C2—N1 −177.28 (11) C2—C1—C6—C5 −0.87 (19)
C7—C1—C2—N1 5.44 (19) C7—C1—C6—C5 176.55 (11)
C6—C1—C2—C3 0.76 (18) C6—C1—C7—O1 1.70 (18)
C7—C1—C2—C3 −176.52 (11) C2—C1—C7—O1 179.02 (12)
N1—C2—C3—C4 178.14 (11) C6—C1—C7—O2 −177.87 (11)
C1—C2—C3—C4 0.01 (19) C2—C1—C7—O2 −0.55 (19)
C2—C3—C4—C5 −0.8 (2) C3—C4—N2—O4 179.95 (12)
C2—C3—C4—N2 −179.65 (11) C5—C4—N2—O4 1.00 (19)
C3—C4—C5—C6 0.67 (19) C3—C4—N2—O3 0.1 (2)
N2—C4—C5—C6 179.56 (11) C5—C4—N2—O3 −178.89 (13)
C4—C5—C6—C1 0.16 (19)
Symmetry code: (i) −x, −y+2, −z+1.
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
N1—H1A···O2 0.88 (2) 2.04 (2) 2.6935 (17) 130.8 (18)
N1—H1B···O1ii 0.82 (2) 2.12 (2) 2.9258 (17) 167.8 (17)
N3—H3A···O5iii 0.90 (2) 1.95 (2) 2.7960 (18) 157.5 (19)
N3—H3B···O3iv 0.87 (2) 2.41 (2) 3.0564 (18) 130.9 (17)
N3—H3B···N1v 0.87 (2) 2.52 (2) 3.1847 (18) 133.8 (18)
N3—H3C···O2ii 0.917 (19) 1.937 (19) 2.8250 (17) 162.5 (14)
O5—H5A···O1ii 0.84 (2) 1.91 (2) 2.7429 (16) 179 (2)
O5—H5B···O2vi 0.79 (3) 2.18 (3) 2.9647 (16) 171 (3)
C6—H6···O1 0.93 2.41 2.7454 (16) 101