organic papers
o930
Eriksson and Hu C12H7Br3O DOI: 10.1107/S1600536801014702 Acta Cryst.(2001). E57, o930±o932Acta Crystallographica Section E
Structure Reports Online
ISSN 1600-5368
Phenyl 2,4,6-tribromophenyl ether
Lars Erikssona* and Jiwei Hub
aDivision of Structural Chemistry, Arrhenius-laboratory, Stockholm University, S-106 91 Stockholm, Sweden, andbDepartment of Chemistry, University of JyvaÈskylaÈ, FIN-40 351 JyvaÈskylaÈ, Finland
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study
T= 293 K
Mean(C±C) = 0.006 AÊ
Rfactor = 0.033
wRfactor = 0.065
Data-to-parameter ratio = 16.7
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2001 International Union of Crystallography Printed in Great Britain ± all rights reserved
The title compound, C12H7Br3O, is the third well characterized
of a total of 209 different brominated diphenyl ethers. The bromine-substituted rings pack in the crystal in a common plane with the same ring from symmetry-related neighbouring molecules. The short Br Br contact distance [3.519 (2) AÊ], together with a pair of considerably longer Br Br contact distances [3.966 (2) AÊ], may be part of a model describing this packing.
Comment
One of the most important groups of ¯ame retardants is that of the polybrominated diphenyl ethers (PBDE). There are 209 possible brominated congeners, but most of the commercially available mixtures consist of highly brominated congeners such as decabromodiphenyl ether (Eriksson et al., 1999). Brominated diphenyl ethers are additive ¯ame retardants, which means that they are only mixed together with plastic material and therefore they migrate more easily to the environment than if they had been covalently bonded with the polymer material (Kuryla & Papa, 1979). The number of known PBDEs without any heterosubstituent, such as hydroxylsetc., is rather limited. In the autumn 2001 release of the Cambridge Structural Database (CSD; Allen & Kennard, 1993), only three PBDE's are listed and one of these without coordinates. Including hetero substituents other than bromine gives a larger set of structures for use as model compounds, but still only of the order of 10±15 structures. One salient feature of the PBDEs is that they are often not found in the environment in proportion to their use. A possible reason for this is that they are decomposed by sunlight, radicals or some other reactions in the environment (OÈrnet al., 1996). It is a long-term goal to try to model the reactivity of different PBDEs that are often found deposited on solid soot particles
etc. Thus an accurate model of the crystal structure is impor-tant.
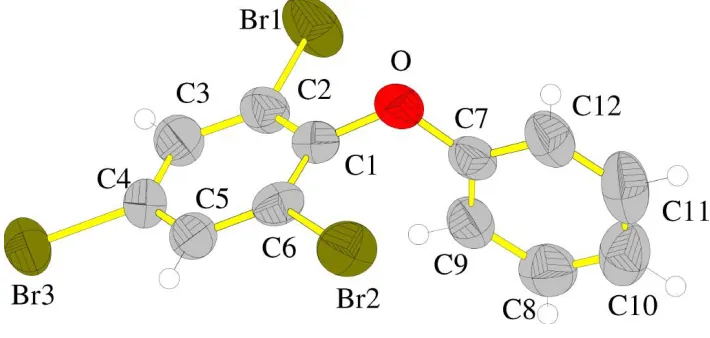
The packing of the title compound, (I) (Fig. 1), shows some interesting features. The brominated ring (C1±C6) is planar within 0.005 AÊ, with Br1 deviating by 0.034 (6) AÊ, Br2 in the
ring plane, Br3 deviating by 0.036 (5) AÊ and the O atom deviating by 0.084 (5) AÊ from the ring plane; the other benzene ring is planar within <0.01 AÊ, also with the O atom in the plane. The angle between the ring planes is 89.8 (1). The
molecules pack in such a way that bromine-substituted rings from different molecules all occupy the same plane (Fig. 2).
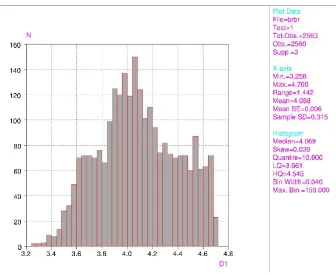
The C atoms of the constituent Br-substituted rings deviate by <0.01 AÊ from the ring plane. At least one of the Br contacts, Br2 Br2(ÿx+ 2, ÿy+ 1, ÿz+ 1), is rather short at 3.519 (2) AÊ, while the next longer contact, Br3 Br1(ÿx, ÿy+ 2, ÿz+ 1), is 3.966 (2) AÊ. The ®rst of these Br Br contacts is considered short compared with similar distances from a search of all intermolecular Br Br distances in Br-substituted aromatic compounds in the CSD (Allen & Kennard, 1993), shown in Fig. 3. Whether the packing of the molecules in the crystal is an effect of the interhalogen bonding or if the short halogen contacts are consequences of the packing in the crystal is an open question.
Experimental
The synthesis of the title PBDE was carried out by coupling the diphenyliodonium salt with a bromophenylate (Beringeret al., 1959; Ziegler & Marr, 1962; Hu, 1996, 1999). The title compound was recrystallized from methanol.
Crystal data
C12H7Br3O Mr= 406.91
Triclinic,P1
a= 5.994 (2) AÊ
b= 10.182 (5) AÊ
c= 11.479 (5) AÊ
= 109.43 (5)
= 99.76 (4)
= 97.37 (5)
V= 638.1 (3) AÊ3
Z= 2
Dx= 2.118 Mg mÿ3
MoKradiation Cell parameters from 1255
re¯ections
= 1.7±25.0
= 9.46 mmÿ1 T= 293 (2) K Irregular, colourless 0.240.150.13 mm
Data collection
Stoe IPDS diffractometer Area-detector scans
Absorption correction: numerical (X-RED; Stoe & Cie, 1997)
Tmin= 0.104,Tmax= 0.303
9866 measured re¯ections 2425 independent re¯ections
1701 re¯ections withI> 2(I)
Rint= 0.055 max= 25.9 h=ÿ7!7
k=ÿ12!12
l=ÿ14!14
Re®nement
Re®nement onF2 R[F2> 2(F2)] = 0.033 wR(F2) = 0.066 S= 1.23 2425 re¯ections 145 parameters
H-atom parameters constrained
w= 1/[2(F
o2) + (0.02P)2]
whereP= (Fo2+ 2Fc2)/3
(/)max= 0.001
max= 0.59 e AÊÿ3
min=ÿ0.38 e AÊÿ3
Table 1
Selected geometric parameters (AÊ,).
Br1ÐC2 1.884 (4) Br2ÐC6 1.894 (4) Br3ÐC4 1.907 (4)
OÐC1 1.381 (4) OÐC7 1.394 (4) C1ÐOÐC7 117.3 (3)
OÐC1ÐC6 121.7 (4) OÐC1ÐC2 119.7 (3)
C9ÐC7ÐO 123.2 (3) C12ÐC7ÐO 115.5 (4) C7ÐOÐC1ÐC6 78.7 (4)
C7ÐOÐC1ÐC2 ÿ107.1 (4) C1ÐOÐC7ÐC9C1ÐOÐC7ÐC12 ÿ155.5 (3)26.2 (5)
Data collection: EXPOSE in IPDS Software (Stoe, 1997); cell re®nement:CELLinIPDS Software; data reduction:INTEGRATE in IPDS Software; program(s) used to solve structure: SHELXS97 (Sheldrick, 1990); program(s) used to re®ne structure:SHELXL97
Acta Cryst.(2001). E57, o930±o932 Eriksson and Hu C12H7Br3O
o931
organic papers
Figure 1
The molecule of the title compound with the atom-numbering scheme. Displacement ellipsoids are shown at the 50% probability level. H atoms are shown as small circles of arbitrary radii.
Figure 2
Packing diagram of the title compound [symmetry codes: (#1)ÿx+ 2, ÿy+ 1,ÿz+ + 1; (#2)ÿx,ÿy+2,ÿz+ 1].
Figure 3
organic papers
o932
Eriksson and Hu C12H7Br3O Acta Cryst.(2001). E57, o930±o932(Sheldrick, 1997); molecular graphics: DIAMOND (Bergerhoff, 1996).
This work was supported by a grant from the Swedish Natural Science Research Council.
References
Allen, F. H. & Kennard, O. (1993).Chem. Des. Autom. News,8, 1, 31±37. Bergerhoff, G. (1996).DIAMOND. Gerhard-Domagk-Straûe 1, 53121 Bonn,
Germany.
Beringer, F. M., Falk, R. A., Karniol, M., Lillien, G., Masullo, M., Mausner, M. & Sommer, E. (1959).J. Am. Chem. Soc.81, 342±351.
Eriksson, J., Eriksson, L. & Jakobsson, E. (1999).Acta Cryst.C55, 2169±2171.
Hu, J. (1996). Polybrominated Diphenyl Ethers (PBDE) Synthesis and Characterization. Licenciate Thesis, Department of Environmental Chem-istry, Stockholm University, Sweden.
Hu, J. (1999).Peristent Polyhalogenated Diphenyl Ethers: Model Compound Syntheses, Characterization and Molecular Orbital Studies. Doctorate Thesis, Department of Chemistry, Research Report No. 73, University of JyvaÈskylaÈ, Finland.
Kuryla, W. C. & Papa, A. J. (1979).Flame Retardancy of Polymeric Materials, Vol. 5. New York: Dekker.
OÈrn, U., Eriksson, L., Jakobsson, E. & Bergman, AÊ (1996).Acta Chem. Scand.
50, 802±807.
Sheldrick, G. M. (1990).Acta Cryst.A46, 467±473.
Sheldrick, G. M. (1997).SHELXL97. University of GoÈttingen, Germany. Stoe & Cie (1997).IPDS Software(Version 2.87, December 1997) andX-RED
supporting information
sup-1
Acta Cryst. (2001). E57, o930–o932supporting information
Acta Cryst. (2001). E57, o930–o932 [doi:10.1107/S1600536801014702]
Phenyl 2,4,6-tribromophenyl ether
Lars Eriksson and Jiwei Hu
S1. Comment
One of the most important group of flame retardants are the polybrominated diphenyl ethers (PBDE). There are 209
possible brominated congeners, but most of the commercially available mixtures consists of highly brominated congeners
such as decabromodiphenyl ether (Eriksson et al., 1999). Brominated diphenyl ethers are additive flame retardents, which
means that they are only mixed together with the plastic material and therefore they migrate more easily to the
environment than if they had been covalently bonded with the polymer material (Kuryla & Papa, 1979). The number of
known PBDEs without any heterosubstituent as hydroxyls etc. are rather limited. In the autumn 2001 release of the
Cambridge Structural Database (CSD; Allen & Kennard, 1993), only three PBDE's are listed and one of these without
coordinates. Including other hetero substituents than bromine gives a larger set of structures for use as model compounds,
but still only in the order of 10–15 structures. One salient feature of the PBDEs is that they are often not found in the
environment to the same extent as used. A possible reason for this is that they are decomposed by sunlight, radicals or
some other reactions in the environment (Örn et al., 1996). It is a long-term goal to try to model the reactivity of different
PBDEs that are often found deposited on solid soot particles etc. Thus an accurate model of the crystal structure are
important.
The packing of the title compound, (I), shows some interesting features. The brominated ring (C1—C6) is planar within
0.005 Å, with Br1 deviating by 0.034 (6) Å, Br2 within the ring plane, Br3 deviating by 0.036 (5) Å and the O atom
deviating by 0.084 (5) Å from the ring plane; the other benzene ring is planar within <0.01 Å, also with the O atom in the
plane. The angle between the ring planes is 89.8 (1)°. The molecules pack in a way that bromine-substituted rings from
different molecules all occupy the same plane (Fig. 2). The C atoms of the constituent Br-substituted rings deviate by
<0.01 Å from the ring plane. At least one of the Br contacts, Br2···Br2(-x + 2,-y + 1,-z + 1), is rather short at 3.519 (2) Å,
while the next longest contact, Br3···Br1(-x,-y + 2,-z + 1), is 3.966 (2) Å. The first of these Br···Br contacts is considered
short compared with similar distances from a search of all intermolecular Br···Br distances in Br-substituted aromatic
compounds in the CSD (Allen & Kennard, 1993), shown in Fig. 3. Whether the packing of the molecules in the crystal is
an effect of the interhalogen bonding or if the short halogen contacts are consequences of the packing in the crystal is an
open question.
S2. Experimental
The synthesis of the title PBDE was carried out by coupling the diphenyliodonium salt with a bromophenylate (Beringer
supporting information
[image:5.610.127.482.75.244.2]sup-2
Acta Cryst. (2001). E57, o930–o932Figure 1
The molecule of the title compound with the atom-numbering scheme. Displacement ellipsoids are shown at the 50%
probability level. H atoms are shown as small circles of arbitrary radii.
Figure 2
[image:5.610.124.480.323.472.2]supporting information
[image:6.610.144.480.79.354.2]sup-3
Acta Cryst. (2001). E57, o930–o932Figure 3
Histogram of intermolecular Br···Br distances where the Br substitutes an aromatic ring. Data are from the CSD autumn
2001 release (Allen & Kennard, 1993).
Phenyl 2,4,6-tribromophenyl ether
Crystal data C12H7Br3O
Mr = 406.91
Triclinic, P1 a = 5.994 (2) Å b = 10.182 (5) Å c = 11.479 (5) Å α = 109.43 (5)° β = 99.76 (4)° γ = 97.37 (5)° V = 638.1 (3) Å3
Z = 2 F(000) = 384 Dx = 2.118 Mg m−3
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 1255 reflections θ = 1.7–25.0°
µ = 9.46 mm−1
T = 293 K
Irregular, colourless 0.24 × 0.15 × 0.13 mm
Data collection Stoe IPDS
diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
Detector resolution: 6.0 pixels mm-1
area–detector scans
Absorption correction: numerical (X-RED; Stoe & Cie, 1997) Tmin = 0.104, Tmax = 0.303
9866 measured reflections 2425 independent reflections 1701 reflections with I > 2σ(I) Rint = 0.055
θmax = 25.9°, θmin = 3.4°
supporting information
sup-4
Acta Cryst. (2001). E57, o930–o932Refinement Refinement on F2
Least-squares matrix: full R[F2 > 2σ(F2)] = 0.033
wR(F2) = 0.066
S = 1.23 2425 reflections 145 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H-atom parameters constrained w = 1/[σ2(F
o2) + (0.02P)2]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max = 0.001
Δρmax = 0.59 e Å−3
Δρmin = −0.38 e Å−3
Special details
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes)
are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2,
conventional R-factors R are based
on F, with F set to zero for negative F2. The threshold expression of
F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R
-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be
even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
Br1 0.07262 (9) 0.73606 (6) 0.15501 (4) 0.08616 (19)
Br2 0.77796 (8) 0.57412 (5) 0.44077 (4) 0.07397 (16)
Br3 0.28026 (9) 0.97473 (5) 0.68889 (4) 0.07956 (18)
O 0.4696 (5) 0.5877 (3) 0.2044 (2) 0.0617 (7)
C1 0.4208 (7) 0.6706 (4) 0.3159 (3) 0.0513 (9)
C2 0.2538 (7) 0.7518 (4) 0.3119 (3) 0.0558 (10)
C3 0.2126 (7) 0.8431 (4) 0.4224 (3) 0.0622 (10)
H3 0.1000 0.8976 0.4195 0.075*
C4 0.3426 (7) 0.8514 (4) 0.5372 (3) 0.0563 (10)
C5 0.5095 (7) 0.7737 (4) 0.5446 (3) 0.0566 (10)
H5 0.5949 0.7805 0.6228 0.068*
C6 0.5486 (6) 0.6846 (4) 0.4330 (3) 0.0520 (9)
C7 0.3932 (7) 0.4412 (4) 0.1640 (3) 0.0528 (10)
C8 0.1430 (9) 0.2331 (5) 0.1495 (4) 0.0745 (12)
H8 0.0165 0.1901 0.1699 0.089*
C9 0.2065 (8) 0.3802 (4) 0.1957 (3) 0.0610 (11)
H9 0.1241 0.4362 0.2471 0.073*
C10 0.2649 (10) 0.1507 (5) 0.0742 (4) 0.0842 (15)
H10 0.2232 0.0521 0.0450 0.101*
C11 0.4488 (10) 0.2142 (6) 0.0422 (4) 0.0856 (15)
H11 0.5300 0.1580 −0.0100 0.103*
supporting information
sup-5
Acta Cryst. (2001). E57, o930–o932H12 0.6390 0.4020 0.0635 0.081*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
Br1 0.0885 (4) 0.1222 (4) 0.0522 (2) 0.0472 (3) 0.0070 (2) 0.0321 (2)
Br2 0.0660 (3) 0.0895 (3) 0.0816 (3) 0.0366 (2) 0.0182 (2) 0.0420 (2)
Br3 0.1070 (4) 0.0740 (3) 0.0548 (2) 0.0298 (3) 0.0220 (2) 0.0135 (2)
O 0.076 (2) 0.0645 (17) 0.0588 (14) 0.0225 (14) 0.0345 (14) 0.0280 (13)
C1 0.057 (3) 0.051 (2) 0.0513 (19) 0.0105 (18) 0.0153 (17) 0.0234 (16)
C2 0.060 (3) 0.063 (2) 0.0479 (18) 0.0191 (19) 0.0093 (17) 0.0238 (17)
C3 0.069 (3) 0.066 (2) 0.059 (2) 0.028 (2) 0.0152 (19) 0.0248 (19)
C4 0.066 (3) 0.047 (2) 0.052 (2) 0.0110 (19) 0.0148 (19) 0.0124 (16)
C5 0.065 (3) 0.051 (2) 0.0488 (19) 0.0068 (19) 0.0033 (18) 0.0176 (17)
C6 0.047 (2) 0.053 (2) 0.066 (2) 0.0131 (17) 0.0138 (18) 0.0313 (18)
C7 0.059 (3) 0.065 (3) 0.0409 (17) 0.021 (2) 0.0109 (17) 0.0241 (17)
C8 0.075 (3) 0.076 (3) 0.064 (2) −0.001 (2) 0.004 (2) 0.027 (2)
C9 0.067 (3) 0.065 (3) 0.0482 (19) 0.017 (2) 0.0126 (19) 0.0154 (18)
C10 0.095 (4) 0.067 (3) 0.067 (3) 0.017 (3) −0.011 (3) 0.008 (2)
C11 0.085 (4) 0.088 (4) 0.065 (3) 0.041 (3) 0.003 (3) 0.001 (2)
C12 0.068 (3) 0.082 (3) 0.053 (2) 0.030 (2) 0.018 (2) 0.019 (2)
Geometric parameters (Å, º)
Br1—C2 1.884 (4) C5—H5 0.9300
Br2—C6 1.894 (4) C7—C9 1.371 (5)
Br3—C4 1.907 (4) C7—C12 1.382 (5)
O—C1 1.381 (4) C8—C10 1.368 (7)
O—C7 1.394 (4) C8—C9 1.388 (6)
C1—C6 1.382 (5) C8—H8 0.9300
C1—C2 1.383 (5) C9—H9 0.9300
C2—C3 1.383 (5) C10—C11 1.372 (7)
C3—C4 1.382 (5) C10—H10 0.9300
C3—H3 0.9300 C11—C12 1.373 (6)
C4—C5 1.362 (5) C11—H11 0.9300
C5—C6 1.379 (5) C12—H12 0.9300
C1—O—C7 117.3 (3) C9—C7—C12 121.3 (4)
O—C1—C6 121.7 (4) C9—C7—O 123.2 (3)
O—C1—C2 119.7 (3) C12—C7—O 115.5 (4)
C6—C1—C2 118.3 (3) C10—C8—C9 120.6 (5)
C1—C2—C3 120.9 (3) C10—C8—H8 119.7
C1—C2—Br1 120.2 (3) C9—C8—H8 119.7
C3—C2—Br1 118.9 (3) C7—C9—C8 118.7 (4)
C4—C3—C2 118.5 (4) C7—C9—H9 120.7
C4—C3—H3 120.8 C8—C9—H9 120.7
C2—C3—H3 120.8 C8—C10—C11 119.6 (5)
supporting information
sup-6
Acta Cryst. (2001). E57, o930–o932C5—C4—Br3 119.7 (3) C11—C10—H10 120.2
C3—C4—Br3 118.1 (3) C10—C11—C12 121.0 (4)
C4—C5—C6 118.1 (3) C10—C11—H11 119.5
C4—C5—H5 120.9 C12—C11—H11 119.5
C6—C5—H5 120.9 C11—C12—C7 118.7 (5)
C5—C6—C1 121.9 (4) C11—C12—H12 120.7
C5—C6—Br2 119.0 (3) C7—C12—H12 120.7
C1—C6—Br2 119.1 (3)
C7—O—C1—C6 78.7 (4) O—C1—C6—C5 176.1 (3)
C7—O—C1—C2 −107.1 (4) C2—C1—C6—C5 1.8 (6)
O—C1—C2—C3 −175.5 (4) O—C1—C6—Br2 −5.8 (5)
C6—C1—C2—C3 −1.1 (6) C2—C1—C6—Br2 179.9 (3)
O—C1—C2—Br1 6.1 (5) C1—O—C7—C9 26.2 (5)
C6—C1—C2—Br1 −179.5 (3) C1—O—C7—C12 −155.5 (3)
C1—C2—C3—C4 0.1 (6) C12—C7—C9—C8 1.2 (6)
Br1—C2—C3—C4 178.5 (3) O—C7—C9—C8 179.4 (3)
C2—C3—C4—C5 0.4 (6) C10—C8—C9—C7 0.2 (6)
C2—C3—C4—Br3 −178.8 (3) C9—C8—C10—C11 −1.3 (7)
C3—C4—C5—C6 0.2 (6) C8—C10—C11—C12 0.9 (7)
Br3—C4—C5—C6 179.4 (3) C10—C11—C12—C7 0.4 (7)
C4—C5—C6—C1 −1.3 (6) C9—C7—C12—C11 −1.5 (6)