Outlook
for the
Child
With
a Cephalocele
Mark S. Brown, MD, and Margaret Sheridan-Pereira, FRCPI, DObst DCH, DStats
ABSTRACT. Specific information on the outcome for a
child with a cephalocele can be difficult to find and
interpret. To update outcome information for the child
with a cephalocele, the investigators compared outcome
of 34 infants from their institution with that of those in
previously published series. For the infants from the
investigators’ institution, overall mortality was 29% and
was confined to infants with posterior defects, which was
consistent with other published series. Additional major
congenital abnormalities were present in nearly half the
infants, and these were an important factor in
contrib-uting to a poorer prognosis as well as whether the defect
could be operatively reduced. Seizures and
hydrocepha-lus were often secondary problems in those infants who
did worse. In addressing outlook for the infant with the
cephalocele, primary factors to be considered are
opera-bility and the presence of additional major abnormalities,
both mtracranialand extracranial. Pediatrics
1992;90:914-919; cephalocele, meningocele, encephalocele.
A cephalocele is a congenital prolapse of central nervous system elements, including meninges (men-imgocele) and often brain tissue (encephalocele), through a skull defect.’ Cephaloceles, also termed cranium bifidum, most often occur along the midline of the skull from the anterior to the posterior cranium
and are grouped within the developmental category
of neural tube defects, which also include spina bifida and amemcephaly.
Surgical repair of cephaloceles was first attempted more than 100 years ago, and in 1943 Ingraham and Swan5 reported the first large series of 84 cases of cephalocele, 63 of which were posterior. Posterior defects had a worse prognosis, attributed to hydro-cephalus. With the introduction of ventricular shunt-ing, mortality with cephaloceles decreased to about
45%67 and in the series reported by Lorber7 in 1967
of 55 infants with cephaloceles, the single most im-portant prognostic factor was whether there was brain tissue in the cephalocele sac. Microcephaly was com-mon and associated with both severe mental
retar-dation and physical handicaps. A later report by
Lorber and Schofield8 in 1979 included 347 infants with posterior defects treated between 1954 and 1977, with a similar mortality of 47%; of the 78 surviving infants, only 4% (n = 2) of those with encephaloceles
were normal compared with 48% (n 14) with
From the Department of Perinatology, Children’s Hospital, and Department of Pediatrics, University of Colorado Health Sciences Center, Denver. Received for publication Apr 8, 1992; accepted Jun 1 7, 1992.
Reprint requests to (M.S.B.) P/SL Medical Center, 1719 E 19th Ave. Denver, CO 80218.
PEDIATRICS (ISSN 0031 4005). Copyright © 1992 by the American Acad-emy of Pediatrics.
meningoceles. In this large series, outlook was worse when there was brain tissue in the sac, microcephaly, or a larger defect.
The only series of anterior defects with detailed
follow-up was by Suwanwela and Hongsprabhas9 in
1966. Mortality was only 12%, and 57% of those
examined (n = 14) were normal. Mealey et al’#{176}and
Simpson et a!2 have reported children with anterior
cephaloceles in combined series, with similar findings of a better outlook for anterior defects.
The aim of the current report was to obtain as specific information as possible on prognostic factors for the child with a cephalocele through review of our institution’s experience and that available in the literature. The outlook for the child with a cephalocele primarily depended on whether the defect could be reduced and the impact of additional major abnor-malities.
METHODS
All admissions to The Children’s Hospital in Denver between January 1, 1940, and December 31, 1987, were reviewed, and a total of 34 infants with a cephalocele were identified. Information obtained from the hospital records included maternal history, sex, location and size of the defect, brain tissue involvement, additional abnormalities, surgical treatments, mortality, morbidity, and fol-low-up information. The classification of defects and outcome used was similar to that used by others.23’#{176} Both meningoceles and encephaloceles were included. An encephalocele included any
de-fect that had radiographic or ultrasound imaging showing brain
tissue or had brain resected during surgery. Anterior cephaloceles
were sited at some point between the bregma and the margin of
the ethmoid bone. Posterior cephaloceles were defined as panetal when the defect was between the bregma and the lambda, and
occipital when the defect arose between the lambda and the
foramen magnum, possibly including one or more of the cervical
vertebrae. Size was taken as the greatest measured dimension. One
child had primary surgery done elsewhere and had been admitted for revision. His follow-up information was included.
For follow-up information, contact was made with the parent(s) and primary and specialty physicians. Because of the diversity of information sources, developmental handicaps were categorized into mental, motor, and sensory disabilities for simplicity. When these were present they were graded as either mild or severe, depending on their impact on the child’s functional capacity.
Statistical comparison was by x2.
RESULTS
There were 22 posterior defects (five meningoceles) and 12 anterior defects (eight meningoceles) (Fig 1). Acranium was classified as a posterior defect. There were no cases of basal (nonvisible) anterior cephalo-celes. Five of the anterior defects presented in the midline, and 20 of the posterior cephaloceles were in
the midline. Defects varied in size from 1 cm to 30
cm. The child with the largest defect, which only
Fig 1. Distribution of cephaloceles separated into meningoceles (unshaded) and encephaloceles (shaded).
Anterior
Defects
Posterior
Defects
outcome with only a mild fine motor handicap. There were 1 7 boys and 1 7 girls. The sex distribution ac-cording to the site of the defect was not significantly different; 9 boys and 13 girls had posterior defects, and 8 boys and 4 girls had anterior defects (P = .23).
Maternal history included a mean age 25.7 years and 4 multigravid mothers, 2 of whom had had previous perinatal losses (an unexplained stillborn and an anencephalic infant); 10 had a history of spontaneous abortion; 2 had insulin-dependent diabetes; and 1 mother reported having a cephalocele herself at birth, although these records were not reviewed.
Additional major abnormalities were present in 15 (44%) of 34 infants (Table 1), 7 (58%) with anterior
defects and 8 (36%) with posterior defects. Those
with anterior defects more often had abnormalities of or within the cranium, while those with posterior defects more often had abnormalities in extracranial
sites. Only 3 of the 22 posterior defects (and none of
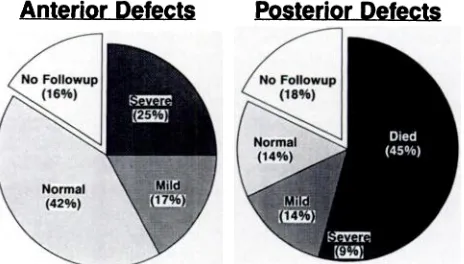
the anterior ones) were considered inoperable. These 3 infants died by 6 months of age. Follow-up infor-mation was available for 28 (82%) of the cases (Fig 2). Age at follow-up was from 1 to 13 years; the 6 without any follow-up were born before 1964 and the families could not be located. For those infants born after 1964, follow-up information was available. The factors that were primarily associated with poorer outcome were posterior location, presence of other major congenital abnormalities, and inoperability.
Mortality was 29% (10/34) and confined to infants with posterior cephaloceles. These deaths (n) were
attributed to complex congenital heart disease (3),
defect mnoperability (3), acranium (1), acute brain herniatiom due to a blocked shunt in a child with mild motor defect (1), and infection in a child with severe handicaps (1); a child with meningomyelocele and severe delays died after the 1 7th hospital admission at 2 years of age (1). Developmental outcome was not associated with either the size of the defect, which ranged from 1 to 30 cm, or the presence or absence of brain tissue in operable defects. In the children
TABLE 1. Additional Major Congenital Abnormalities and Outcome According to Site of Cephalocele
Abnormality Outcome’
Anterior
Cleft lip/palate, NF
craniosynostosis
Myelomeningocele, NF
hydrocephalus
Hydrocephalus Severe developmental delay (8 y) Agenesis of corpus callosum, Severe developmental
anophthalmia, multiple fa- delay (10 y)
cial abnormalities
Porencephaly, anophthalmia, Severe developmental
multiple abnormalities delay (7 y)
Facial clefts, dermoid of corpus Normal (4 y)
callosum
Facial clefts, coloboma Normal (2 y)
Posterior
Complex congenital heart din- Died by 6 mo
ease, hydronephrosis
Complex congenital heart dis- Died at 14 mo
ease, visceral heterotaxia
Complex congenital heart dis- Died at 4 mo
ease, cleft lip/palate, micro-phthalmia
Arhinocephaly, cerebellar Inoperable, died agenesis, hydrocephalus,
pituitary agenesis, adrenal hypoplasia
Myelomeningocele Died at 2 y
Hydrocephalus
()t
2, mild (2 y, 7 y); 1, severe (2 y); developmental delaysKlippel-Feil, hydrocephalus Severe developmental delays (1 y)
Acranium Died at 4 d
0 NF, no follow-up; age at follow-up is given in parentheses.
t(n).
Fig 2. Pie graphs of the outcome of cephaloceles according to their
distribution between anterior and posterior sites.
with anterior defects, severe handicaps were present
in 4 (33%), and mild in 2 (17%) (Table 2). The
presence of other intracranial abnormalities in 2 in-fants with anterior cephaloceles, with or without fa-cial clefting, was associated with severe handicaps. In the surviving children with posterior defects, severe
handicaps were present in 2 (33%), and mild in 3
(50%). Microcephaly was noted in 7 of the children, all with posterior defects; 2 had no follow-up infor-mation, 3 died (all of whom were inoperable), 1 had severe handicaps, and the remaining 1 was normal.
Nine children had hydrocephalus diagnosed and a
TABLE 2. Outcome According to Site of the Cephalocele’
Location Died Severe Handicap Mild Handicap Normal No Follow-up
Anterior
Encephalocele (n = 4)
Meningocele (n = 8)
...
...
1, Severe mental and motor (8 y) 2, Severe mental,
motor, and visual (13 y, 7 y)
1, Mild motor; 1, mild visual (12 y)
3, Normal (2 y, 3 y, 4 y)
2, Normal (2 y, 3 y)
..
2
Posterior
Encephalocele (n = 17) 9 1, Severe mental,
motor, and blind
(1 y)
1, Mild fine motor and strabismus
(7 y); 1, mild
memory deficit (lOy)
2, Normal (4#{189}y,
5 y)
3
Meningocele (n = 5) 1 (No repair due to heart defect)
1, Severe mental and motor (4’/2 y)
1, Mild perceptual and fine motor (5
y)
1, Normal (5 y) 1
Totals (n = 34) 10 5 5 8 6
0 Age at follow-up is given in parentheses.
posterior ones. One of these died, 5 had severe hand-icaps, 1 had mild delays, 1 was normal, and 1 had no follow-up information. Seizures were recorded in 14 children, 5 with anterior defects and 9 with posterior defects; of these, 5 died, 5 had severe handicaps, 1 had mild handicaps, 1 was normal, and 2 were unable to be followed up.
DISCUSSION
The outlook for the child with a cephalocele pri-manly depended on site, operability, and additional major abnormalities. Overall mortality was 29%, but confined to those with posterior defects and associ-ated with inoperability, congenital heart disease, in-fection, and shunt malfunction. Microcephaly, sei-zures, and hydrocephalus occurred in almost half of the infants, and when present, were secondarily as-sociated with a worse outcome. The association of congenital abnormalities was different between the posterior and anterior defects, but with either, addi-tional major abnormalities were an important consid-eration in determining the infant’s prognosis. This information represents an expanded view of the out-look for the child with a cephalocele.
Although the frequency of cephaloceles has ranged
from 1 per 240011 to 1 per 12 500 live births,’2 most
estimates have been between 1 per 5000 and 1 per
9000.1321 Prevalence more aptly applies to the re-ported frequency of cephaloceles because the true incidence is hidden in stillborns, underreferral of
mas-sive defects, and early pregnancy losses. This latter
has been estimated to be from 7%22 to 20%’ of all cephalocele defects. Since our series is from a regional referral center, it is likely that this misrepresents the
true
population prevalence, because not all infants may have been referred, especially those with ob-viously inoperable defects, which are more often pos-terior. There is considerable geographic variation in the proportions of anterior23and posterior’5.’#{176}’7’31M cephaloceles, although overall prevalence is about thesame.19’203’ Posterior defects were 65% of our series,
which falls into the range of reports from the Western hemisphere.
Meningoceles comprised 38% of our defects, a
fig-ure similar to the 37% in the combined series of
Simpson et al.2 Meningoceles included 67% and 29%
of anterior and posterior defects, respectively, in our
infants. In series of posterior defects, meningoceles
include 18% to 49% of the defects,6’7 while in those
series of anterior defects this distinction has not been
made.
The sex distribution of anterior defects varies widely, from 40%9b0 to 73% females.27’35 In our series, females were 33% of those with anterior defects. The sex distribution of infants with posterior defects has ranged from 5#{216}%68lo.18 to
75%
females.’2 In ourseries, 62% of the posterior defects were in females. A female predominance in livebirths with cephalo-celes has been speculated to be due to an inordinately
high early prenatal loss of male fetuses with
cepha-loceles,36 although from the considerable variation in
sex distribution in the above reports this does not seem to always hold true.
The site of the inner bony defect in the skull of
anterior cephaloceles has been localized to the region
of the foramen cecum.3’26’31’37’38 The facial component of the defect determines the subclassification.38 The most common anterior defect was the nasofrontal cephalocele, which presents at the glabella and whose external opening is between the frontal and nasal bones.3’23’29 The site of posterior defects is most often
along the median plane, as were 9 1
%
of those in ourseries. The level varies and similarly determines the subclassification. When the defect is between the parietal bones, the defect is called parietal; when it is at or around the occipital prominence it is termed occipital, suboccipital when it occurs below this, and occipitocervical when it extends into the cervical re-gion.39 The association of Klippel-Feil deformity with
posterior cephaloceles and the similarity between
in-iencephaly and Klippel-Feil syndrome suggests that cephaloceles are related to iniencephaly and spina bifida.4 Posterior defects located below the occipital
prominence may extend into the cervical region and
are frequently associated with other intracranial ab-normalities of the posterior fossa. When defects in-volve the occipital protuberance, a venous sinus
usu-ally runs in the neck of the sac.27 Large occipital
defects may extend both above and below the
por-tions of cerebrum and cerebellum with absence of the
tentorium.3 These factors not only make surgery
dif-ficult or impossible as in three of our cases, but ma also affect both long-term morbidity and mortality.2
The etiology of the cephalocele defect is unclear.
Environmental and genetic factors have been
exam-med and both seem to play a role.”9”2’20’2225’35’4#{176} Other etiologic considerations have arisen from ani-mal studies in which cephaloceles have been pro-duced by folic acid antagonists, trypan blue, vitamin A excess, malnutrition, and x-irradiation.28 A recent
double-blind trial of preconception folic acid
supple-mentation in women with a previous child with a
neural tube defect demonstrated a significant
reduc-tion in neural tube defects, although cephalocele was
not reported separately.4’
The cephalocele abnormality occurs between 25
and 50 days for anterior defects and up to 60 days for posterior ones.4’42 The association of cephaloceles with an abnormality in heart development, which is
occurring at the same time, suggests that the two
occurring together may represent a field defect. Other
major congenital abnormalities, both intracranial and extracranial, were present in 44% of our infants,
which is within the range of previous reports.3’8’20’43
Cohen and Lemire44 reviewed more than 20
syn-dromes associated with cephaloceles, which in our series included amniotic band syndrome, absent
cor-pus callosum, facial clefting, Klippel-Feil, and
men-ingomyelocele.4548 Cephaloceles associated with the amniotic band syndrome are clearly a separate etio-logic sequence and the facial clefts are characteristi-cally multiple and asymmetric.47 Three of the children with anterior defects in our series had significant
facial clefting characteristic of the amniotic band
syn-drome including involvement of the eyes.
Associated abnormalities with anterior defects are more often limited to the cranium.9’31 In our series these included cleft lip and palate, porencephaly,
anophthalmia, facial hypoplasia, agenesis of corpus
callosum, and dermoid of corpus callosum.
Micro-cephaly occurred in 7% to 24% of the infants in
Suwanwela and Hongsprabhas’ series,9 although we did not find any record of microcephaly in those with
anterior defects. This may have been due to the
smaller number of children with anterior defects in
our series, differences in measurement of head cir-cumference, or a different association of other abnor-malities in populations with a higher prevalence of anterior defects. From 12% to 24% of infants with
anterior defects have had hydrocephalus,9’3’ which is
consistent with the 1 7% in our series. Significant eye
abnormalities occurred in 25% of those with anterior
defects, in accord with other reports.9’28 Defects in brain development have been reported to occur in 3% to 23% of infants with anterior defects,9’24’26’28
al-though intracranial abnormalities have not been
rou-tinely assessed in any series. We found 25% of our infants with anterior defects had some identified
ab-normality of the brain. In an autopsy series of 12
children with anterior defects, Suwanwela and
Suwanwela37 reported brain abnormalities of
holo-telencephaly, abnormal gyri, hydrocephalus, small frontal lobe, and elongated brain stem.
In the infants with posterior defects in our series, other major congenital abnormalities occurred in 25%. We found one infant with Klippel-Feil
syn-drome, the most frequently identified condition
as-sociated with posterior cephaloceles.43 There were two infants with myelomeningoceles in our series, one with an anterior and the other with a posterior defect. On occasion, cephaloceles have been observed within families also affected with spina bifida, hydro-cephalus, or other central nervous system
anoma-lies.3’8 However, the infrequent association between
cephaloceles and neural tube defects in most series
would suggest that these two entities are not closely
related.7’8 In those with posterior defects, congenital
abnormalities were more likely to be extracranial and
included congenital heart disease, limb abnormalities,
hydronephrosis, visceral heterotaxia, asplenia, and
myelomeningocele. In the literature, associated
ab-normalities have included congenital heart disease, limb abnormalities, hydrocephalus, microcephaly, cleft lip and palate, and tracheoesophageal fistula. Hydrocephalus was present in 18% of our infants with posterior defects. Hydrocephalus has been re-ported to occur with posterior cephalocele in 16% to 5 1 % of patients, without distinction between
menin-gocele and encephalocele.26’8’#{176}’33 The association of
cephalocele with other congenital abnormalities,
ex-cluding hydrocephalus and microcephaly, ranges
from 1 7% to 61
%,
often including both minor andmajor abnormalities grouped together.8’41’43 These
es-timates are probably underestimates, inasmuch as
evaluations have not been systematic and those in-fants deemed inoperable have died early and were often excluded.
Outcome
Outcome for the child with an anterior defect has been universally better than for those with a posterior defect.33 Survival with anterior defects has been
re-ported from 80% to 93%#{149}9.b0.24.29 Those with posterior
cephaloceles in our series had a 55% survival, com-parable with the 40% to 75% survival reported in
other series.2’7’8”0 In our series, mortality was most
often due to the impact of other abnormalities or the
inability to safely repair the defect. All babies with lesions deemed inoperable died within 6 months. Meningoceles have had a much lower mortality-25% in our infants with posterior defects and 10% in Lorber and Schofield’s 1979 series.8
Disabilities in children surviving with anterior cephaloceles have been more frequent in those with encephaloceles, although this was not our experience, which may have been due to our smaller numbers or a different definition of encephalocele. The primary
morbidity in children with anterior defects has been
facial disfigurement, anosmia, and visual prob-lems.9”#{176}Although there were no deaths in our infants
with anterior defects, significant long-term disability
was present in 50% of those who had follow-up and was associated with severe facial clefting, intracranial abnormalities, and significant visual impairments. Factors that did not seem to influence outcome in-cluded sex, presence of brain tissue in the defect, and
without brain tissue, the important factor in deter-mining outcome was the presence of other intracra-nial abnormalities.
In infants with posterior defects, handicaps have been related to brain abnormalities, growth retarda-tion, Klippel-Feil deformity, ataxia, blindness,
hydro-cephalus, fine motor coordination, and seizures.4
Se-vere long-term disabilities occurred in 25% and mild handicaps in 38% of the surviving infants with
fol-low-up in our series, regardless of the presence or
absence of brain tissue or size of the defect when
operable. This is possibly explained by the anatomy of the remaining underlying brain, which may be as
important a factor in determining outcome as the
amount of prolapsed tissue. This is supported by 41% of the infants with seizures, of whom 71 % died or had severe handicaps. In other reports, disabilities have ranged from 14% to 73%, the variability de-pending on the extent and details of follow-up.2’7’10
The parietal location of posterior defects is far less
frequent and accounted for 9% of our posterior
de-fects, within the 3% to 27% in the literature.1’3’5”0’32
A series by McLaurin49 made a detailed study of 13 children with parietal encephaloceles. Mortality was 3 1 % and limited to those with brain tissue within the
defect. Follow-up ranged from 9 months to 10 years,
and 5 of the surviving 9 were considered normal.
Seven of the 1 3 had meningoceles, of which 4 were
normal, and none died. In this location,
encephalo-celes rather than meningoceles are associated with
intracranial abnormalities such as agenesis of the
corpus callosum, upward extension of the third
yen-tide, Dandy-Walker c’sts, or porencephalic cysts of the lateral ventricles.3’4
Predictors of Outcome
Although the number of infants seen in our
insti-tution with cephalocele was less than those in other
reports, the distribution of site, sex, mortality, and
outcome was comparable with earlier reports. Those
infants with an anterior defect not associated with
facial clefting or brain abnormalities had good
out-comes. In the infants with posterior defects, poor
outcome was related to other major congenital
ab-normalities, infection, and inoperabiity. In our series
as well as others, hydrocephalus and seizures were a secondary consideration of poor outcome. Inopera-bility is an end of the spectrum of considerations enumerated by Lorber for poor prognosis with
pos-terior cephalocele, which includes the triad of
mi-croencephaly, presence of brain tissue, and large size.7’8 In our series if the defect was able to be safely
closed, then microcephaly, size, and brain tissue
in-volvement did not alone worsen the outlook. This may be due to improvements in diagnostic
evalua-tions, surgical techniques, and postoperative intensive
care. Of interest was that in follow-up in the later
report, 48% of infants with a meningocele were fully
normal compared with only 4% with encephaloceles.8 The infants in our series with posterior encephalocele had the highest mortality but when they survived did not have any worse outcome.
In the report by Simpson et al,2 of 74 patients
treated in Australia between 1955 and 1983, the
prognostic importance of presence of brain tissue and size of herniation was emphasized. These authors
divided the size at 5 cm, with only 8% of those with
smaller lesions doing poorly, compared with 75% of
those with larger lesions. In the series reported by
Mealey et al,’#{176} prognosis depended largely on
whether the lesion contained cerebellum. The size of
lesions ranged from 1 to 14 cm,1#{176}and those children
who were normal in follow-up had a maximum defect
diameter of 7 cm. In our series, size alone was not
associated with poorer outcome since not only did a
number of children with smaller defects do worse,
but some of the larger defects contained only a small
amount of brain tissue. Lesions that have been
deemed inoperable have universally done poorly, with a high mortality.8’10 The posterior defects that were considered inoperable in our series were greater
than 7 cm and contained all brain tissue. Those with
large defects that only partially contained brain were
operable, and these children had a better outcome.
Prenatal Diagnosis
Antenatal screening with maternal serum and
am-niotic fluid a-fetoprotein has not been of much value
because of the closed nature of most cephaloceles.2
Cephaloceles have been diagnosed antenatally by
ultrasound.2’50’5’ In a screening series reported by
Winsor and St John Brown16 from Nova Scotia, none
of the cephalocele defects were missed beyond 16
weeks in the latter part of the study, and for the entire
period from 1980 through 1984, 79% of either spina bifida or cephaloceles were diagnosed antenatally. Anterior defects are less often diagnosed antenatally
because of their lower prevalence in the screened
populations reported, their smaller sizes, and
diffi-culties with technique.52 The differential diagnosis of a cephalocele seen on antenatal ultrasound includes
other central nervous system defects such as dermoid
cyst, hydrocephalus, Dandy-Walker cyst, or a cystic
hygroma of the neck.51 The recommendation of late
termination of pregnancy needs to be with the
exclu-sion of other similar-appearing abnormalities and the
unlikelihood of operability based on posterior
loca-tion, larger size, presence of microcephaly, and
pri-mary inclusion of brain tissue in the sac.5#{176} Summary
In summary, the practitioner caring for an infant
with a cephalocele can use the following to address
the initial care, referral, and counseling of the family.
Associated abnormalities are present in up to 50% of the cases. Mortality depends on cephalocele location, operability, and additional major abnormalities. The presence of brain tissue in the defect does not
neces-sarily
bode a poor prognosis unless the defect is so large that it is considered inoperable. Additionaleval-uation may include computed tomographic scan,
The option of nonintervention in the care of an infant with a cephalocele can be ethically supported in the infant with microcephaly and a massive defect con-taming all or almost all brain tissue, a condition that is deemed inoperable. In addition, the infant with a larger defect and other significant major congenital abnormalities may have operative risks outweighing surgical benefit.
ACKNOWLEDGMENT
The cooperation of all the parents was deeply appreciated.
REFERENCES
1. David JD, Proudman TW. Cephaloceles: classification, pathology, and
management. World JSurg. 1989;13:349-357
2. Simpson DA, David JD, White J. Cephaloceles: treatment, outcome and antenatal diagnosis. Neurosurgery. 1984;15:14-21
3. Nager GT. Cephaloceles. Laryngoscope. 1987;97:77-84
4. Warkany J, Lemire RJ, Cohen MM. Encephaloceles. In: Warkany J, Lemire RJ, Cohen MM, eds. Mental Retardation and Congenital
Malfor-mations of the Central Nervous System. Chicago, IL: Year Book Medical
Publishers Inc; 1981:158-175
5. Ingraham FD, Swan H. Spina bifida and cranium bifidum, I: a survey of five hundred and forty-six cases. N Engl JMed. 1943;228:559-563 6. Guthkelch AN. Occipital cranium bifidum. Arch Dis Child. 1970;45:
104-109
7. LorberJ. The prognosis of occipital encephalocele. Dcv Med Child Neurol.
1967;9(suppl B):75-86
8. Lorber J, Schofield JK. The prognosis of occipital encephalocele. Z
Kinderchir. 1979;28:347-351
9. Suwanwela C, Hongsprabhas C. Fronto-ethmoidal encephalomenin-gocele. I Neurosurg. 1966;25:172-182
10. Mealey J, Dzenitis AJ, Hockey AA. The prognosis of encephaloceles. I.
Neurosurg. 1970;32:209-218
1 1. Leck I, Record RG, McKeown T, Edwards JH. The incidence of
malfor-mations in Birmingham, England, 1950-1959. Teratology. 1968;1:263-280
12. Sever LE. An epidemiologic study of neural tube defects in Los Angeles County, II: etiologic factors in an area with low prevalence at birth.
Teratology. 1982;25:323-334
13. Myrianthopoulos NC. Our load of central nervous system malforma-tions. In: Myrianthopoulos NC, Bergsma D, eds. Recent Advances in the
Developmental Biology of Central Nervous System Malformations. New
York, NY: Alan R. Liss Inc; 1979;1-18
14. Hams LE, Steinberg AG. Abnormalities observed during the first six
days of life in 8716 live-born infants. Pediatrics. 1954;14:314-326
15. Haase J, Green A, Hauge M, Holm NV, Mathiasen H. A cohort study of neural tube defects (NTD) in Denmark covering the first seven years of life. Childs Nero Syst. 1987;3:117-120
16. Wmsor EJT, St John Brown B. Prevalence and prenatal diagnosis of neural tube defects in Nova Scotia in 1980-84. Can Med Assoc J.1986;
135:1269-1273
17. Field B. The child with an encephalocele. Med IAust. 1974;1:700-703 1 8. Field B. Neural tube defects in New South Wales, Australia. JMed Genet.
1978;15:329-338
19. Khoury Mi, Erickson JD, Cordero JF, McCarthy BJ. Congenital malfor-mations and intrauterine growth retardation: a population study.
Pedi-atrics. 1988;82:83-90
20. Wiswell TE, Tuttle DJ, Northam RS, Simonds GR. Major congenital neurologic malformations. AJDC. 1990;144:61-67
21. Edmonds LD, James LM. Temporal trends in the incidence of malfor-mation in the United States in selected years, 1970-71, 1982-83. Centers for Disease Control. CDC Surveillance Summaries. 1985;34 (No. 255):1-3
22. Sever LE, Sanders M, Monsen R. An epidemiologic study of neural tube defects in Los Angeles County, I: prevalence at birth based on multiple sources of case ascertainment. Teratology. 1982;25:315-321
23. Suwanwela C. Geographical distribution of fronto-ethmoidal encepha-lomeningocele. BrJ Prey Soc Med. 1972;26:193-198
24. Whatmore WJ. Sincipital encephalomeningoceles. Br J Surg. 1973;60:
261-270
25. Thu A, Kyu H. Epidemiology of frontoethinoidal encephalomeningocele in Burma. JEpidemiol Community Health. 1984;38:89-98
26. Naiin-Ur-Rahman. Nasal encephalocele. I Neurol Sci. 1979;42:73-85
27. Tandon PN. Meningoencephaloceles. Acta Neurol Scand. 1970;46: 369-383
28. Rapport RL II, Dunn RC Jr, Aihady F. Anterior encephalocele. I
Neuro-surg.1981;54:213-219
29. De Kierk DJJ, De villiers JC. Frontal encephaloceles. SAft Med I. 1973; 47:1350-1355
30. Onuigbo WIB. Encephaloceles in Nigerian Igbos. JNeurol Neurosurg
Psychiatry. 1977;40:726-730
31. Fisher RG, Uihlein A, Keith HM. Spina bifida and cranium bifidum: study of 530 cases. Proc StaffMtg Mayo Clin. 1952;27:33-38
32. Lipschitz R,Beck JM, Froman C, Phil D. An assessment of the treatment of encephalomeningoceles. S Aft MedJ. 1969;24:609’-#{243}lO
33. Odeku EL. Congenital malformations of the cerebrospinal axis seen in
Western Nigeria: the African child with ‘encephalocele.’ mt Surg. 1967;
48:52-62
34. Matson DD. Cranium bifidum and encephalocele. In: Matson DD, ed.
Neurosurgery of Infancy and Childhood. Springfield, IL: Charles C
Thomas; 1969:61-75
35. David DJ, Sheffield L, Simpson D, White J. Fronto-ethmoidal meningo-cephaloceles: morphology and treatment. Br I Plast Surg. 1984;37: 271-284
36. Creasy MR. Alberman ED. Congenital malformations of the central nervous system in spontaneous abortions. IMed Genet. 1976;13:9-16
37. Suwanwela C, Suwanwela N. A morphological classification of sincipital encephalomeningoceles. JNeurosurg. 1972;36:201-21 I
38. David DJ, Simpson DA. Frontoethmoidal meningocephaloceles. Clin
Plast Surg. 1987;14:83-89
39. O’Rahilly R. Anomalous occipital apertures. Arch Pathol. 1952;53: 509-519
40. Janench DT. Endocrine dysfunction and anencephaly and spina bifida: an epidemiologic hypothesis. Am JEpidemiol. 1974;99: 1-6
41. MRC Vitamin Study Research Group. Prevention of neural tube defects: results of the medical research council vitamin study. Lancet. 1991;338: 131-137
42. Volpe JJ. Human brain development. In: Volpe JJ, ed. Neurology of the
Newborn. Philadelphia, PA: WB Saunders Company; 1987:2-32
43. Hunter AGW. Neural tube defects in Eastern Ontario and Western Quebec: demography and family data. Am JMed Genet. 1984;19:45-63
44. Cohen MM Jr, Lemire RJ. Syndromes with cephaloceles. Teratology.
1982;25:161-172
45. Seeds JW, Cefalo RC, Herbert WNP. Amniotic band syndrome. Am I
Obstet Gynecol. 1982;144:243
46. Sakoda K, Ishikawa 5, Uozumi T, Hirakawa K, Okazaki H, Harada Y. Sphenoethmoidal meningoencephalocele associated with agenesis of corpus callosum and median cleft lip and palate. JNeurosurg. 1979;51: 397-401
47 Yokota A, Matsukado Y, Fuwa I, Moroki K, Nagahiro S. Anterior basal encephalocele of the neonatal and infantile period. Neurosurgery. 1986;
19:468-478
48. Hudgins RJ, Edwards MSB, Ousterhout DK, Golabi M. Pediatric neuro-surgical implications of the amniotic band disruption complex. Pediatr
Neurosci. 1985-86;12:232-239
49. McLaurin RL. Panetal cephaloceles. Neurology. 1964;14:764-772 50. Chatterjee MS. Bondoc B, Adhate A. Prenatal diagnosis of occipital
encephalocele. Am JObstet Gynecol. 1985;153:646-647
51, Pilu G, Rizzo N, Orsini LF, Bovicelli L. Antenatal recognition of cerebral anomalies. Ultrasound Med Biol. 1986;12:319-326
52. Donnenfeld AE, Hughes H, Weiner S. Prenatal diagnosis and perinatal
management of frontoethmoidal meningoencephalocele. Am IPerinatol.
1988;5:51-53
53. Lusk RP, Dunn VD. Magnetic resonance imaging in encephaloceles. Ann