An Approach to Renal Masses
in Pediatrics
Alpin D. Malkan, MDa, Amos Loh, MDa, Armita Bahrami, MDb, Fariba Navid, MDc,d, Jamie Coleman, MDe, Daniel M. Green, MDf, Andrew M. Davidoff, MDa, John A. Sandoval, MDa
abstract
Renal masses in children may be discovered during routine clinical examination or incidentally during the course of diagnostic or therapeutic procedures for other causes. Renal cancers are rare in the pediatric population and include a spectrum of pathologies that may challenge the clinician in choosing the optimal treatment. Correct identification of the lesion may be difficult, and the appropriate surgical procedure is paramount for lesions suspected to be malignant. The purpose of this article is to provide a comprehensive overview regarding the spectrum of renal tumors in the pediatric population, both benign and malignant, and their surgical management.Renal cancers are rare in children, accounting for 6% to 7% of all childhood tumors.1They can be detected by a parent bathing or holding the child, during routine physical examination or screening of children with known clinical
syndromes with predispositions to renal disease,2or incidentally during investigations for other
intraabdominal processes.3–5The key challenge is distinguishing malignant neoplasms from benign masses. A thorough understanding of the profile of common renal masses in children, as well as their associated clinical and imaging features, can facilitate accurate preoperative diagnosis and optimize patient care.
This article comprehensively reviews the approach to identifying renal cancers in children, individually summarizes the history, diagnosis, histopathology, and management of malignant and benign pediatric renal masses (Table 1), and provides the rationale for choice of the correct surgical procedure.
APPROACH
The priority of management is the differentiation of nonneoplastic
processes from benign and malignant neoplasms (Fig 1). A thorough review of the patient’s clinical history and physical examination may reveal additional signs or symptoms to aid in the diagnosis. This may be useful in characterizing nonneoplastic renal pseudotumors, which are masslike imagingfindings that mimic
neoplasms.6The presence offlank pain, fever, or pyuria, for example, is suggestive of an infective process, such as pyelonephritis, instead of a tumor. However, the absence of symptoms neither suggests nor refutes
a diagnosis of malignancy. This will be particularly true of congenital
anomalies such as prominent columns of Bertin or dromedary humps. Although masslike renal lesions in children can sometimes be suspected on plain radiographs and evaluated with ultrasound, subsequent computed tomography (CT) is usually necessary for further characterization and confirmation of the diagnosis.7Several image-based criteria can help to guide the differentiation of malignant from benign lesions.8For solid lesions, once pseudotumors are excluded, the presence of contrast enhancement may be indicative of a neoplasm although Departments ofaSurgery,bPathology,cOncology,eRadiology,
andfEpidemiology and Cancer Control, St. Jude Children’s Research Hospital, Memphis, Tennessee; anddDepartment of Pediatrics, College of Medicine, University of Tennessee Health Science Center, Memphis, Tennessee
Drs Malkan, Loh, Green, Davidoff, and Sandoval conceptualized, designed, wrote, and revised the manuscript; Drs Bahrami, Navid, and Coleman coordinated the collection of histopathologic and radiologic imaging and critically reviewed the manuscript for important intellectual content; and all authors approved thefinal manuscript as submitted.
www.pediatrics.org/cgi/doi/10.1542/peds.2014-1011
DOI:10.1542/peds.2014-1011 Accepted for publication Jul 23, 2014
Address correspondence to John A. Sandoval, MD, Department of Surgery, St. Jude Children’s Research Hospital, MS 133, Room D3045, 262 Danny Thomas Place, Memphis, TN 38105-3678. E-mail: john. sandoval@stjude.org
PEDIATRICS (ISSN Numbers: Print, 0031-4005; Online, 1098-4275).
Copyright © 2015 by the American Academy of Pediatrics
FINANCIAL DISCLOSURE:The authors have indicated they have nofinancial relationships relevant to this article to disclose.
FUNDING:No external funding.
several other abnormalities including infection (pyelonephritis) and abscess require consideration. The claw sign is classically seen as the tumor grows due to the normal kidney being splayed around the tumor and indicative of renal origin.9 CT and especially MRI can
characterize the tissue composition of these solid lesions and can
differentiate soft tissue from fat,fluid, or hemorrhage. On serial imaging, an enlarging mass can spur worries about a neoplasm, although absolute tumor size may not correlate well with malignancy risk.
For cystic masses, the Bosniak renal cyst classification is well established in the literature for malignancy risk prediction in image-detected renal cysts in adults.10Based on this classification system, the lesion’s imaging (originally based on CT
findings, but ultrasound and MRI are commonly applied) morphology and enhancements characteristics are used to categorize each lesion into 1 of 5 groups (I and II are benign and do not require further evaluation; IIF requires follow-up to prove benignity; III are indeterminate and considered surgical lesions although some may prove to be benign; IV are clearly malignant cystic masses that require surgical removal).8,11–13A modified Bosniak classification has been shown to correlate fairly well with
pathologic outcomes of complex renal cysts in children but has not been formally validated.14
IMPLICATIONS FOR BIOPSY
Despite thorough clinical and radiographic evaluation, some renal masses will remain indeterminate, and their management is subject to individual clinical opinions. Careful correlation of clinical and imaging
findings may facilitate the
preoperative diagnosis of most renal lesions. Although histologic
evaluation is the gold standard for pathologic diagnosis, obtaining tissue biopsy via percutaneous (using Tru-cut biopsy, open biopsy, orfi ne-needle aspiration) or open surgical approach can have serious
implications that require consideration. If a renal mass is suspicious for malignancy, the multidisciplinary team in addition to the surgeon typically determines the potential for complete
resectability of the primary mass, and whether there is any evidence of metastatic disease on preoperative imaging. If the mass is determined to be unresectable then the issue of biopsy becomes more apparent to guide therapy.
In children, general anesthesia is typically needed to perform either an open or percutaneous biopsy. An open biopsy requires an incision, allows for large specimens to be obtained as compared with a percutaneous needle approach; however, this method inevitably causes the patient more discomfort, increased recovery time, and scarring. Even more problematic is obtaining an inadequate sample with potential for sampling error. In many cases, a percutaneous needle biopsy cannot differentiate renal lesions such as Wilms tumor (WT) from hyperplastic nephrogenic rests.15More disturbing are the consequences of upstaging the tumor, which is used to describe a patient’s cancer from a lower stage (less extensive) to a higher stage (more extensive),15necessitating administration of additional chemotherapeutic agents and/or radiation therapy. For instance, in
FIGURE 1
Renal mass algorithm.
TABLE 1 Renal Lesions in Children
Benign Malignant
Angiomyolipomas WT
Renal pseudotumor Clear cell sarcoma Renal cell carcinoma MA
Clear cell
Multicystic nephroma Medullary carcinoma Metanephric stromal
tumor
Multicystic
ORTI Chromophil
Reninoma Chromophobe
Collecting duct Papillary Translocation
Xp11.2 t(6;11) Nephroblastomatosis Rhabdoid tumor Anaplastic sarcoma Congenital mesoblastic
nephroma DSRCT
Ewing sarcoma/PNET Synovial sarcoma
lower stage WT without anaplasia, complete resection of the mass, along with adjuvant chemotherapy, results in cure rates of over 90% at 5 years, whereas resection alone results in 20% long-term survival rate.16,17 A preoperative needle biopsy in this particular scenario would upstage the malignancy, and the possible need for more aggressive treatment, including chemotherapy and radiation.
An emphasis on the multidisciplinary approach is essential to the
evaluation and treatment of these patients including the pediatrician, pediatric radiologist, pediatric surgeon, pediatric urologist, pediatric oncologist, pediatric radiation oncologist, and pathologist. The key characteristics of a comprehensive spectrum of pediatric renal masses are outlined below.
MALIGNANT NEOPLASMS
Wilms tumor
WT, also called nephroblastoma, is the most common pediatric renal malignancy,18–21accounting for∼6% to 7% of all childhood renal
cancers,18and is observed at a frequency of 1:10 000 newborn infants,18,22occurring mostly
between 2 and 5 years of age.19Most cases are sporadic, although it can occur in hereditary form with autosomal dominant inheritance, variable penetrance, and several affected generations.18Symptoms may include abdominal pain, distension, and hypertension; however, the patient can also be asymptomatic. Bilaterality occurs in 4% to 13% of cases. WT may be associated with genitourinary anomalies and some congenital syndromes such as WAGR, Denys-Drash, and Beckwith-Wiedemann syndromes (BWS).15
WAGR syndrome includes WT, aniridia (absence of the colored part of the eye), genitourinary
abnormalities (gonadal tumors), and mental retardation.15Interstitial
deletion on chromosome 11p13 (prevalence of 0.4% of children with WT),15,23and bilateral WT (15%) may be seen in association with WAGR syndrome.15,24 Denys-Drash syndrome is characterized by the triad of WT, pseudohermaphroditism, and chronic renal failure. Individuals with Denys-Drash syndrome manifest early nephrotic syndrome, which progresses to diffuse mesangial sclerosis and subsequent renal failure usually within thefirst 3 years of life. Germline missense mutations in the WT1gene are responsible for most WTs that occur as part of the Denys-Drash syndrome25including the 90% risk of developing WT.15,26BWS includes macroglossia, macrosomia (birth weight and length greater than the 90th percentile), midline
abdominal wall defects (omphalocele, umbilical hernia, diastasis recti), ear creases or ear pits, and neonatal hypoglycemia. It is also characterized by the development of WT
(bilateral),27rhabdomyosarcoma, and hepatoblastoma.15BWS results from constitutional loss of imprinting or heterozygosity ofWT2.15
Approximately 1 of 5 patients with BWS and WT may present with bilateral disease; however,
metachronous bilateral disease is also observed.15,28–30
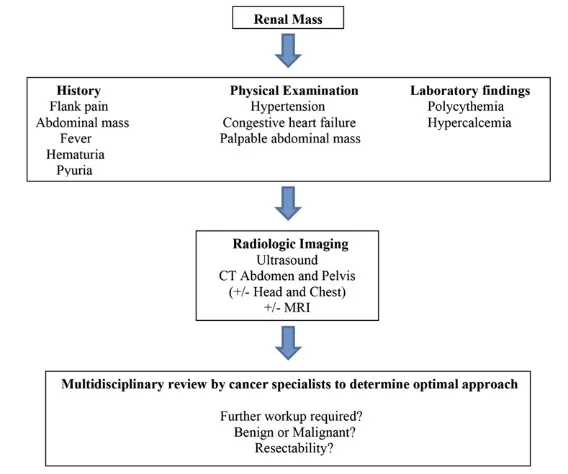
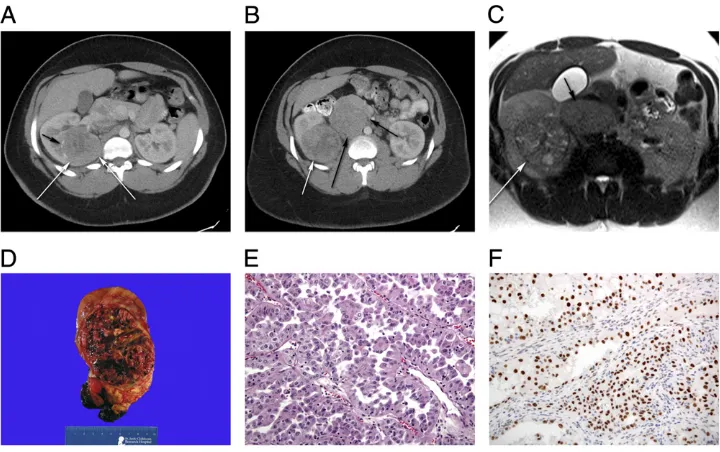
WT typically appears as a large, smooth, well-defined intrarenal mass with uniform echogenicity that often displaces neighboring structures. WT is usually seenfirst on ultrasound, and subsequently with less enhancement on CT than adjacent normal
parenchyma (Fig 2).7Calcifications, fat density, patches of renal parenchymal enhancement, invasion of the renal collecting system, vascular invasion with tumor thrombus extension into the renal vein and inferior vena cava (most accurately evaluated on MRI), or the presence of distant lung metastases may be seen.7Rarely, it may be largely cystic, an entity referred to as cystic partially differentiated nephroblastoma, that cannot be differentiated from cystic
nephroma on imaging, but outcomes of both are favorable regardless of treatment.31Current imaging modalities may fail to detect occult synchronous contralateral tumors at the time of initial diagnosis, but these occur in only a very small percentage of patients with WT, and outcomes for these patients have also been
excellent.32
Preoperative staging studies in patients suspected of having WT include CT (or MRI) scans of the chest, abdomen, and pelvis.15The stage is determined by the results of imaging studies, as well as surgical and pathologicfindings at time of nephrectomy, and is the same for tumors with favorable or anaplastic histology. In stage I (43% of patients) and II (20%) WTs, the tumor is completely resected without evidence of tumor at or beyond the margins of resection.15Stage III (21%) and IV (11%) WTs have either residual nonhematogenous tumor present after surgery that is confined to the abdomen or hematogenous
metastases or lymph node metastases outside the abdominopelvic region are present, respectively.15Lymph node and major blood vessel involvement (encasement and/or thrombus) are reflective of advanced, potentially unresectable disease and higher stage (III or IV).15
clusters that are considered to increase the risk for tumor formation in the remaining kidney.15
Unilateral WT is generally treated with nephrectomy followed by adjuvant chemotherapy. Neoadjuvant chemotherapy may be used to promote tumor shrinkage and is used in bilateral (Fig 3) or inoperable WTs and in selected treatment protocols. Postoperative radiation therapy may be administered to the tumor bed for local control, or to the whole abdomen in cases of gross spillage or dissemination. Metastatic disease and unfavorable histology are poor prognostic factors in WT.15 Children with WAGR syndrome or other germline WT mutations require monitoring throughout their lifetime because of the increased risk of developing hypertension, nephropathy, and renal failure.15,33Current
screening recommendation’s for children with WAGR, Denys-Drash, and BWS or any genetic predisposition
(overgrowth syndrome, sporadic aniridia, or isolated hemihypertrophy) that increases the chance of developing WT include abdominal ultrasounds every 3 months until 8 years of age.15,28,34–36Approximately 5% to 10% of individuals with WT have bilateral or multicentric tumors, and those with genetic predisposition have an increased prevalence.15Eighty-five percent of patients with either WAGR or BWS have unilateral tumors.15,37 Because∼10% of patients with BWS will develop a malignancy, most commonly WT or hepatoblastoma, screening with abdominal ultrasound in addition to seruma-fetoprotein is recommended until age 4 (because most hepatoblastoma occur before this age), with renal ultrasounds
thereafter.38Currently, there is no standard approach for the treatment of those predisposed to developing bilateral WT.
Patients with Denys-Drash syndrome are faced with a significant challenge regarding the controversial need for
early prophylactic bilateral nephrectomies as a means to diminish the potential development of WT,39–44the adverse effects of chemotherapy, including the
prolongation of time to consideration for transplantation, and to reduce metabolic and nutritional sequelae of chronic renal failure.40Some authors propose careful monitoring of patients every 4 to 6 months by abdominal ultrasonography and performing nephrectomy after the onset of end-stage renal failure45,46; however, others feel it is preferable to proceed with bilateral nephrectomies“early” rather than“late”in the course of the underlying renal disease.40
Clear Cell Sarcoma of the Kidney
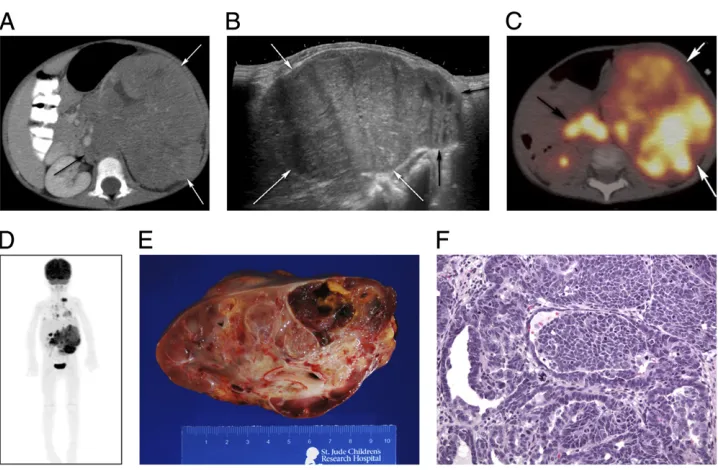
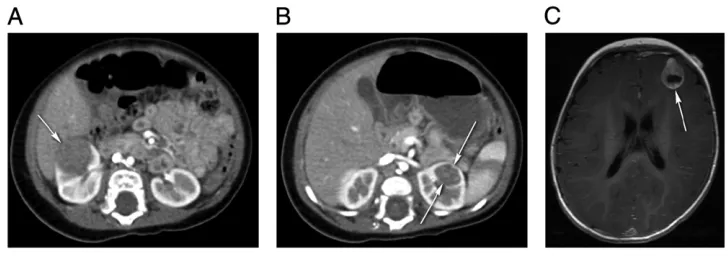
Clear cell sarcoma of the kidney (CCSK) is the second most common renal tumor in children, accounting for 3% to 5% of all childhood cancers.47–50It commonly appears in children younger than 4 years of age.18,22,51CCSK is an aggressive FIGURE 2
tumor with a unique predilection for bone and brain metastasis,20,22,49,51 but can also spread to the lung and abdomen. Symptoms may include abdominal pain, hypertension, and hematuria. Typical presentation includes a large, unilateral, well-circumscribed, sharply demarcated mass49that compresses the surrounding normal renal parenchyma and displaces the collecting system7(Fig 4). CCSK is described as an enigmatic tumor type because its morphologic appearance does not resemble that of the kidney.52It can mimic and be mimicked by WT, congenital
mesoblastic nephroma, and rhabdoid tumor of the kidney.18
The classic microscopic pattern includes cords of round or spindle-shaped cells with clear cytoplasm and ovoid to rounded vesicular nuclei with inconspicuous nucleoli.47,52The cells are surrounded byfibrovascular septa ranging from a thin“chicken-wire” arrangement to broad sheets containing an arborizing capillary vasculature.47,52Cytogenetic abnormalities involving recurring translocations in t(10;17)(q22;p13/ p12)53and deletion of 14q24q31 have been described in CCSK.18Staging according to the National Wilms Tumor Study Group (NWTS) 5 definition demonstrated that stages I to III are nearly equally represented (25%, 37%, 34%, respectively), whereas 4% present with stage IV and evidence of metastatic disease.47Approximately
29% of CCSK cases may have evidence of lymph node metastases.47Ipsilateral renal hilar lymph nodes are the most common site of metastases and underscore the importance of lymph
node sampling in staging. Current multimodal treatment consists of radical nephrectomy for resectable tumors followed by intensive chemotherapy and radiotherapy.49,54 FIGURE 3
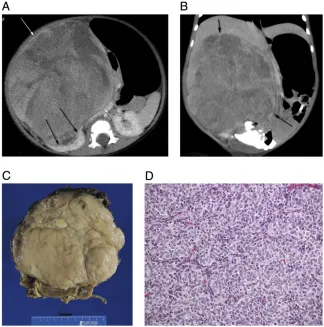
Bilateral WT. A, Axial short TI inversion recovery (STIR) MRI sequence through the upper abdomen demonstrates large heterogeneous masses (black arrows) arising from both kidneys. Note the compressed renal parenchyma (white arrows). B, Axial and (C) coronal STIR MRI sequences of the abdomen demonstrate the size of the large heterogeneous bilateral renal masses, which essentiallyfill the abdomen, and half-Fourier acquisition single-shot turbo spin-echo MRI sequence clearly reveals the small cystic areas within the masses.
FIGURE 4
The addition of doxorubicin to the treatment protocol has led to actuarial 6-year survival approaching 98% for localized disease.47
Renal Cell Carcinoma
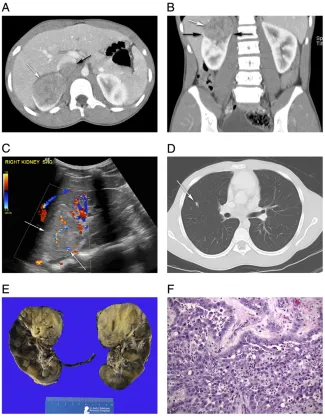
Renal cell carcinoma (RCC) accounts for,0.3% of all childhood tumors,55 is the most common renal tumor in adolescents,56and the average age of presentation is approximately 10–11 years old.57,58RCC is a malignancy thought to arise from epithelial cells of the renal tubule.57Several histologic subtypes of RCC exist, including clear-cell and medullary carcinoma (Fig 5), multicystic,59 chromophil, chromophobe,60and collecting duct.2,61,62Papillary and translocation, Xp11.263(Fig 6) or the t(6;11), types are more common in children.18,64,65RCCs are seen with greater frequency in childhood cancer survivors55,66,67and in genetic syndromes such as von Hippel-Lindau disease, tuberous sclerosis,20,55,68familial clear-cell renal cancer, hereditary papillary renal carcinoma, hereditary
leiomyomatosis, and renal cell cancer syndrome, in individuals with cystic or end-stage renal diseases,69,70 sickle cell hemoglobinopathies, and in pediatric kidney transplant recipients.61
Unlike WT, RCC is rarely
asymptomatic (only 12% of cases).56 Diagnostic clues may include the classic triad of gross, painless hematuria,flank pain, and the presence of a palpable mass.20,56,71,72 Metastases most commonly occur in the lungs (40% to 65%) and bones (10% to 42%)57; however, the liver (35% to 57%), bladder, brain, or pleura (7% to 15%) may also be involved.58Abdominal ultrasound with a subsequent CT scan is used to better define the neoplasm.73 Distinguishing between RCC and other renal tumors requires histologic examination, and usually few if any hints come from imaging.56Although on CT, RCC typically reveals a large, heterogeneous, solid mass with either
well-circumscribed or poorly defined borders.7Intravenous enhancement of the tumor is usually less than the adjacent normal parenchyma.7,51 RCC tends to be smaller than WT, invades tissues locally with distortion of normal renal architecture, and formation of a pseudocapsule that contain foci of
calcification. Regional
lymphadenopathy and vascular invasion are commonly seen.7 RCC histology demonstrates epithelial cells of renal tubule origin with moderate to large amounts of clear and variably eosinophilic cytoplasm with nested, solid, acinar, and/or
tubulopapillary architectural growth
FIGURE 5
patterns.74Papillary growth pattern has been associated with advanced stage by some authors although there are no definitive conclusions regarding the role of histopathology on outcome.57 Translocation RCC is immunoreactive forTFE3nuclear protein, and positive by reverse transcription-polymerase chain reaction for theASPL-TFE3fusion transcript.15Additionally, renal medullary carcinoma is associated with sickle cell hemoglobinopathy and is most commonly seen in young adults.7,75Histologically, it displays various morphologies including reticular, yolk sac, or adenoid cystic growth pattern and sometimes reveals neoplastic cells admixed with
neutrophils in a desmoplastic
background.76Loss of heterozygosity at chromosome 22q11 and q12 and the lack of SMARCB1 protein expression are commonly seen.76
Tumor stage appears to be the most important factor for survival.58The 5-year survival for stage I is 90% or higher, for stages II and III it is 50% to
80%, and for stage IV is 9%, which is similar to the stage-for-stage survival in RCC in adults.15Although the therapeutic value of complete retroperitoneal lymph node dissection is still controversial, the presence of lymph node disease is known to significantly worsen the survival of patients with RCC.58The mainstay of treatment remains radical
nephrectomy with regional
lymphadenectomy15,56,71,77; however, nephron sparing surgery has been performed with success.78RCCs are generally resistant to traditional cytotoxic therapies and are poorly responsive to radiotherapy.56 Immunotherapy, such as interferon-a and interleukin-2, may have some effect on cancer control.15,79–81 Several targeted agents (for example, sorafenib,82sunitinib,83
bevacizumab,84temsirolimus,85 pazopanib,86and everolimus87) have been approved for use in adults with RCC; however, these have undergone limited testing in pediatric patients.
Nephroblastomatosis
Closely associated with WT, nephroblastomatosis is an abnormality of renal development characterized by persistence of nephrogenic rests, which are foci of metanephric blastema that persist into infancy.22,51Metanephric blastema is an embryonic structure with cells derived from the
intermediate mesoderm that normally mostly give rise to the nephron.88These premalignant lesions are believed to give rise to 30% to 40% of WT, and in comparison with WT,
nephroblastomatosis is diffuse, reveals heterogeneity, and is usually bilateral. They can be classified as perilobar, which is limited to the periphery of the renal lobe, well-circumscribed and usually multiple, or intralobar, which is singular, poorly circumscribed, and can be found anywhere within the renal lobe.89 They are detected incidentally in 1% of infants and are associated with
FIGURE 6
BWS, hemihypertrophy and Perlman syndrome in their perilobar form, and with Denys-Drash syndrome, WAGR syndrome, and sporadic aniridia in their intralobar form.7
The diagnosis can be made
radiographically and may appear on CT as low-attenuation, multifocal nodules located at the renal periphery with mild contrast enhancement or as diffuse nephromegaly with retention of the kidney’s reniform shape.22MRI is particularly useful for diagnosis of nephroblastomatosis when it demonstrates homogeneity of the hypointense rindlike lesion on contrast enhancement, which differentiates it from WT.15 Microscopicfindings range from benign small nests of blastemal cells to large areas with frank malignant transformation (WT), and is extremely difficult to differentiate between the 2 based on needle or even incisional biopsies alone.90,91Current
recommendations are for treatment with chemotherapy (vincristine and dactinomycin) until nearly complete resolution as determined by imaging.15However, given the high incidence of bilaterality and the subsequent development of WT, renal sparing surgery is indicated (partial nephrectomy).15,89
Rhabdoid Tumor
Rhabdoid tumor of the kidney is a highly aggressive neoplasm accounting for,2% of all pediatric
renal malignancies.18This typically occurs in children younger than 2 years of age and is one of the most lethal, malignant solid tumors in children, with an overall survival rate
,25%.92A distinct clinical presentation with fever, hematuria, young age (mean age 11 months), and high tumor stage at presentation suggests a diagnosis of rhabdoid tumor of the kidney15,93; however, hypercalcemia due to elevated parathormone levels may also be found.7,22 Characteristic CT features include hemorrhage, necrosis, and tumor lobules lined by
calcification.22,51A peripheral subcapsular crescent with
attenuation offluid may be seen in 71% of patients (Fig 7).94
Synchronous or metachronous primary intracranial masses or brain metastases have been established as distinctive features.22Germline mutations inSMARCB1(also known asSNF5orINI1) gene95,96are seen in
∼35%97of patients with rhabdoid tumor consistent with a genetic predisposition.15,98The most distinct histologicfindings are rather large cells with large vesicular nuclei, a prominent single nucleolus, and in some cells, the presence of globular eosinophilic cytoplasmic
inclusions.15,99Treatment modalities include chemotherapy and
nephrectomy; however, current therapy has limited efficacy for this patient population.100,101Four-year
overall survival rates for stages I and II are 42% and stage III and IV are 16%.92
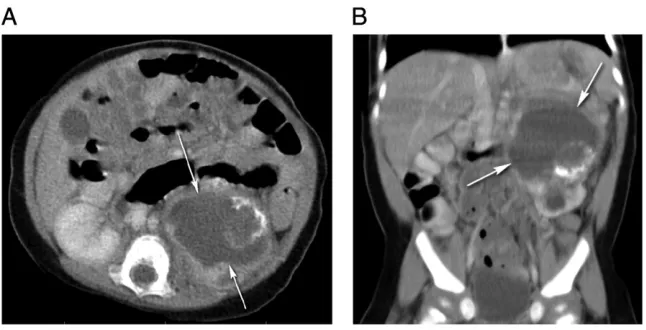
Congenital Mesoblastic Nephroma
Mesoblastic nephroma, the most common solid renal tumor in infancy,20,21,102,103is a low grade fibroblastic sarcoma most commonly affecting infants younger than 3 months, and.90% of cases appear within thefirst year of life.15,18,21The associated manifestations include hypercalcemia with polyuria, congestive heart failure, and hypertension.7Prenatal ultrasound may be helpful because in most cases the diagnosis can be made in the neonatal period as it can lead to polyhydramnios (71% of gestations associated with this tumor), and premature birth.21,104,105A concentric hyperechoic and hypoechoic ring pattern known as the vascular“ring” sign may also be seen surrounding the tumor.106Postnatal CT typically reveals a large, heterogeneous, solid intrarenal mass with smooth margins that enhances to a lesser degree than the adjacent normal renal parenchyma after intravenous contrast
administration (Fig 8).7,104 Based on histopathologic
appearances, congenital mesoblastic nephroma is subtyped into classic (24%), cellular (66%), and mixed (10%) types.103,105,107–109The classic form is morphologically identical to infantilefibromatosis of the renal sinus109with fusiform
FIGURE 7
spindle cells and rare mitoses.105,107 The cellular form is identical to infantilefibrosarcoma,109is characterized by a highly cellular spindle cell proliferation with minimal intervening collagen/ supporting stroma, and high nuclear-to-cytoplasm ratios.108The cellular variant carries a specific
translocation, t(12;15)(p13;q25), involving theETV6andNTRK3genes, similar to the translocation seen in infantilefibrosarcoma.110The mixed type is composed of areas of low (formed by elongated spindle cells resemblingfibroblastic cells seen in benign myofibromas) and high cellularity.108Complete surgical resection via radical nephrectomy is adequate therapy for most patients and reduces the risk of local recurrence.102,105,107In stage III patients (incomplete resection and/ or histologically positive resection margin), or those with cellular subtype, and aged 3 months or older at diagnosis are at increased risk for local and eventually metastatic recurrence. This select group of patients are recommended to have adjuvant chemotherapy.15,107 Five-year event-free survival rate is 94%, and the overall survival rate is 96% when diagnosed within thefirst 7 months of life.111
Anaplastic Sarcoma
Anaplastic sarcoma of the kidney is a recently recognized pediatric tumor112seen in children or
adolescents younger than 15 years of age113with a female predominance.114 Patients typically present with a large renal mass, and the most common sites of metastases are the lung, liver, and bones.15,114The main histologic features are the presence of
undifferentiated spindle cells arranged in short fascicles as well as
undifferentiated small round primitive mesenchymal cells, very marked cellular anaplasia with giant pleomorphic cells, and usually prominent areas of benign or malignant cartilage or chondroid differentiation.114,115Optimal therapy for this tumor is currently unclear.15,114
Other Renal Neoplasms
Several other renal malignant tumors are even less common in children. Desmoplastic small round cell tumor (DSRCT) and Ewing sarcoma/ primitive neuroectodermal tumor (PNET) of the kidney are extremely rare in children.7DSRCT is aggressive, malignant, typically affects children 6 to 8 years of age but may also be seen in young adolescents,18and is diagnosed by its characteristic EWS-WT1translocation.116CT may
reveal a hypovascular, heterogeneous, and well-circumscribed mass with internal punctate calcifications.117It reveals similar histologic features as in the extrarenal sites consisting of nests, cords, or sheets of small
undifferentiated cells with foci of necrosis, calcification, and desmoplasia.115However, the
definitive diagnosis of DSRCT is based on the presence of the typical translocation t(11;22)(p13;q12).18 Patients usually present at an advanced stage with a poor prognosis. Ewing sarcoma/PNETs of the kidney are small round blue cell tumors. The tissue of origin is unknown; however, molecular profiling of these tumors suggests a mesenchymal cell
origin.118–121They are very aggressive, usually large and invasive,19and typically have poorly defined margins with areas of hemorrhage and necrosis.122,123The distinct vascular nature is manifested by an arborizing vascular pattern, perithelial
arrangement of tumor cells, and perivascular pseudorosettes.124More than 90% of renal Ewing sarcoma/ PNETs have characteristic
translocations involving rearrangement of theESWR1gene on chromosome 22, t[11;22][q24;q12].18,124The treatment of these tumors is similar to their counterparts at other sites and includes a combination of chemotherapy, surgery, and/or radiation therapy. Synovial sarcoma of the kidney may present with an abdominal mass andflank pain.115 Histologically they demonstrate a monophasic, spindle cell appearance, with these stromal areas composed of relatively monomorphic, plump, ovoid spindle cells with an intersecting fascicular or solid sheetlike pattern.47 This rare renal tumor is genetically characterized by translocation t[X;18] [p11;q11].18
RENAL TUMOR AS A SECOND MALIGNANCY
In pediatric patients with cancer, renal tumors may be detected on
FIGURE 8
imaging studies conducted during the course of their treatment or during cancer survivorship follow-up surveillance. RCC has been detected as a second malignancy during routine surveillance in survivors of leukemia,66,125supratentorial PNET,66WT,67and
neuroblastoma.55,126In the Childhood Cancer Survivor Study, 51 kidney cancers were detected as second malignancies among 14 359 5-year survivors and another 28 as third or subsequent malignancies.127
SECONDARY INVOLVEMENT OF THE KIDNEY
Primary lymphoma of the kidney is extremely rare.128However, diffuse infiltration of the kidney may be observed in children with leukemia. Involvement of the kidney as seen in non-Hodgkin lymphoma and B-cell lymphomas is usually associated with hematogenous spread or direct extension of retroperitoneal disease.7 On imaging, lymphoma may appear in a variety of forms: as a solitary mass, diffuse nephromegaly, or bilateral multifocal nodules.7The lattermost form may mimic nephroblastomatosis but is unusual in infants and young children. In contrast, leukemic involvement of the kidneys is seen on CT imaging as bilateral diffuse symmetric nephromegaly associated with decreased corticomedullary differentiation and decreased cortical enhancement.51Adjacent or systemic lymphadenopathy is often also visualized on imaging when such renal lesions are detected.
BENIGN NEOPLASMS
Angiomyolipoma
Renal angiomyolipoma is considered a benign mesenchymal tumor, accounts for 3% of solid renal masses,129,130and contains varying amounts of a disordered arrangement of blood vessels, smooth muscle, and adipose tissue.4,22,129Angiomyolipoma may occur sporadically,22,131although
it is seen in up to 80% of patients with tuberous sclerosis (usually bilateral and/or multiple renal
angiomyolipomas).130–132It can be also seen in von Hippel-Lindau syndrome, neurofibromatosis,22Sturge-Weber syndrome, autosomal dominant polycystic kidney disease, and pulmonary lymphangiomyomatosis.133 Presentation at extrarenal sites such as in the liver, spleen, abdominal wall, retroperitoneum, lung, and genital region may also be seen.134Although largely asymptomatic, patients may present withflank pain, fever, nausea, vomiting, hematuria, hypertension, or even retroperitoneal hemorrhage due to aneurysm formation because of the abundant abnormal, elastin-poor vascularity of the tumor.22,130,135 Imaging appearance varies
considerably based on the amount and type of histologic elements present. Diagnosis can usually be made by CT or MRI that reveals macroscopic fat131; however, fat is not pathognomonic or unique to angiomyolipoma and is occasionally seen in WT and RCC.22In fact, radiologic techniques using enhancement patterns and histogram analysis at CT and chemical shift MRI remain controversial to rule out RCC in histologically proven fat poor
angiomyolipomas.136–138A solid renal tumor without fat content on imaging studies is regarded as RCC and treated as such.138
Microscopically, angiomyolipomas are very cellular consisting mainly of myoid cells with vacuolated cytoplasm spinning off of large vessels in a background of mature fat.130In asymptomatic lesions ,4 cm, evaluation with abdominal ultrasound or CT scan should be performed biannually or yearly. In lesions that are.4 cm, symptomatic, or bilateral, arterial embolization and nephron sparing surgery are the treatments of choice130because of the risk of hemorrhage.132
Angiomyolipoma of the kidney tumor remains a benign tumor with rare possibility of transformation to a malignancy139; therefore, follow-up
of these patients remains an important concern.
Follow-up evaluation requirement is proposed for asymptomatic patients with abdominal ultrasound or CT scan of the abdomen (risk of radiation exposure) or MRI at every 6- or 12-month intervals130to monitor the number and size of lesions, and for aneurysm development. The modality used to image children remains a challenge. Ultrasonography is technician dependent, as compared with CT, which requires exposure to radiation, intravenous contrast, and the potential need for general anesthesia. Alternatively, MRI also requires intravenous contrast, with the potential need for general anesthesia and longer time required to complete imaging study. However, the significant advantage of MRI is its detailed imaging, and even more importantly is the elimination of radiation exposure, which makes this more optimal in children. Cost and the availability of these imaging modalities also impact the type of follow-up that may be provided.
Pseudotumors
Renal pseudotumors are masslike imagingfindings (normal renal tissue) that mimic neoplasms.96 These are caused by a variety of conditions including renal congenital anomalies (prominent renal columns of Bertin, dromedary humps), inflammatory masses (focal pyelonephritis, renal abscess, autoimmune disease),7vascular structures (renal artery aneurysm or arteriovenousfistula), or
inflammatory process may include ill-defined margins and perinephric stranding, and abnormalities in the renal vasculature such as vessel hypertrophy may support a diagnosis of vascular malformation. In most cases, a familiarity with this group of masses in addition to a thorough clinical and radiologic evaluation should reveal the correct diagnosis.
Metanephric Adenoma
Metanephric adenoma (MA) is a rare, benign tumor of the renal cortex composed exclusively of epithelial and stromal elements.140–142Patients can be asymptomatic143,144or can have signs and symptoms related to polycythemia, hypertension,
hematuria, dysuria,flank or abdominal pain, or a palpable abdominal mass.3 The lesion has radiologic similarities to RCC and WT, making preoperative diagnosis difficult.141MAs have been diagnosed by using ultrasound141,145 or CT.146Histologic features of MA reveal small uniform epithelial cells of an acinar, tubular, glomeruloid, or papillary growth pattern with a high nuclear-to-cytoplasmic ratio.146 Surgical resection is recommended for MA due to the rarity of the tumor, potential for malignant degeneration, and inability to establish a diagnosis without histologic evaluation141; therefore, nephron-sparing surgery or partial nephrectomy are considered sufficient in most cases.141,147 Although MA is considered benign with a good prognosis,141reports of atypical histologic features, bone metastasis,148and lymphatic dissemination149,150may portend a poor prognosis.151
Cystic Nephroma
Multicystic nephroma is an uncommon, noninheritable, unilateral, benign tumor that represents 2% to 3% of all primary renal tumors in children and is predominantly seen in boys.152,153 Children typically present with a painless, progressively enlarging, palpable abdominal orflank mass that
has a variable growth rate and may be discovered incidentally.152,153Flank pain, urinary tract infection or obstruction, hematuria, and
hypertension may also be seen.154CT may reveal a well-defined,
nonenhancing cystic mass without solid components involving the kidney with multiple enhancing thin internal septa causing compression and stretching of the remaining enhancing renal parenchyma.152The tumor is bulky, well-encapsulated, noninfiltrating, and composed of multiple
noncommunicating,fluid-filled cysts.152 The presence of blastemal cells and poorly differentiated cells must be ruled out to diagnose this lesion. Complete resection either with nephrectomy152or nephron-sparing surgery152,154remains the treatment of choice because carcinomatous
degeneration may occur within the wall of such tumors.152
Metanephric Stromal Tumor
Metanephric stromal tumor is a benign stromal tumor of the kidney seen mostly within thefirst decade of life155with a mean age at
presentation of 2 years.155,156The most common presentation is an abdominal mass.155,156This tumor, MA, and metanephric adenofibroma represent a spectrum of well-differentiated nephroblastic lesions that appear to be related to
WT.142,155,157The most characteristic histologic features include an onion skin ring formation around entrapped tubules and blood vessels with vascular changes, including angiodysplasia/epithelioid transformation of medial smooth muscle and myxoid changes.102It is composed primarily of spindle cells with a low proliferation index, and diffusely infiltrates the perirenal fat.158Nephrectomy is usually curative,102and patients have an overall excellent prognosis.158
Ossifying Renal Tumor of Infancy
Ossifying renal tumor of infancy (ORTI) is a rare, benign renal tumor
that may present as an abdominal mass typically in infants. Boys are more commonly affected, and gross hematuria is almost always observed7,159 due to bulging of the tumor into the pelvicalyceal system.160A soft tissue mass with calcifications near the renal pelvis and no significant contrast enhancement may be seen on CT.7,21,160Histologically, ORTI is characterized by 3 major components: an osteoid core, osteoblastlike cells, and spindle cells.160The best treatment approach for this tumor involves resection6 with kidney preservation
techniques.161
Reninoma
Reninoma, also known as
juxtaglomerular cell tumor, is a rare, benign cause of curable hypertension in children and adolescents.162,163 Patients may be found to have hypertension incidentally, which prompts further investigation leading to the discovery of clinicalfindings including hypokalemia,
hyperaldosteronism, and high renin activity.162,163Reninoma can be divided into typical, atypical, and nonfunctioning types depending on the blood pressure and serum potassium levels.164Although ultrasound may in some
circumstances be helpful, CT and even MRI remain the most useful imaging modalities to document the presence of a reninoma. It may appear isodense or hypodense as compared with the renal
medulla162,163; however, there is no definitive radiologic examination for the diagnosis of reninoma. Therefore, histopathologic evaluation with immunohistochemical staining aids in the correct diagnosis.
Reninoma is composed of polygonal tumor cells within vascular stroma and thick-walled vessels.
Immunohistochemical staining is particularly useful in differentiating reninoma from RCC and
unique to reninoma include the tumor staining positively for CD34, and negatively for CD117, HMB-45, and S100 protein. Because these are usually small and rarely associated with metastasis,166nephron sparing surgery is the standard treatment option allowing for preservation of renal function.162,163
SUMMARY
Upon satisfactory exclusion of renal pseudotumors, renal neoplasms inevitably require some form of intervention to obtain a histologic diagnosis through biopsy or resection. Intervention for these masses is easily justified in view of their significant mass effect and the parental concern that such dramatic lesions would understandably generate. Management decisions need to be tempered with an
understanding of the difference in profile of renal masses in children. Renal masses that are seen within the pediatric population, as discussed here, remain a delicate and sensitive issue. A combination of pathologic rarity, complexity, and uncertain malignant potential may lead to treatment that is either too aggressive, and, in retrospect unnecessary; or that is entirely too conservative when in fact aggressive measures are warranted. We encourage continued advancements in research to better define evidence-based approaches and treatment strategies for these challenging masses.
REFERENCES
1. Howlader N, Noone AM, Krapcho M, et al, eds. SEER Cancer Statistics Review, 1975–2010. Bethesda, MD: National Cancer Institute. Available at: http://seer.cancer.gov/csr/1975_2010/. Accessed October 7, 2014
2. Cameron C, Greenbaum L, Sato T, Trost B, Lundeen B, Kelly ME. Renal cell carcinoma in a patient with cystinosis and inflammatory bowel disease: a case report.Pediatr Nephrol. 2008; 23(7):1167–1170
3. Amodio JB, Shapiro E, Pinkney L, et al. Metanephric adenoma in an 8-year-old child: case report and review of the literature.J Pediatr Surg. 2005;40(5): e25–e28 [Review]
4. Joniau S, Goeman L, Kreuzbauer S, et al. Benign renal mesenchymoma in the pediatric age group: a novel pathologic and karyotype entity.Urology. 2004; 63(5):981–984
5. Akil I, Ozguven A, Canda E, et al. Co-existence of chronic renal failure, renal clear cell carcinoma, and Blau syndrome.Pediatr Nephrol. 2010;25(5): 977–981
6. Sotelo-Avila C, Beckwith JB, Johnson JE. Ossifying renal tumor of infancy: a clinicopathologic study of nine cases. Pediatr Pathol Lab Med. 1995;15(5): 745–762 [Review]
7. Lee EY. CT imaging of mass-like renal lesions in children.Pediatr Radiol. 2007; 37(9):896–907
8. Israel GM, Silverman SG. The incidental renal mass.Radiol Clin North Am. 2011; 49(2):369–383
9. Kim J, Cha S, Daldrup-Link HE, Goldsby R. Pediatric tumors. In: Daldrup-Link HE, Gooding CA, eds.Essentials of Pediatric Radiology: A Multimodality Approach. New York, NY: Cambridge University Press; 2010:204
10. Bosniak MA. The use of the Bosniak classification system for renal cysts and cystic tumors.J Urol. 1997;157(5): 1852–1853
11. Israel GM, Bosniak MA. An update of the Bosniak renal cyst classification system.Urology. 2005;66(3):484–488
12. Bosniak MA. The Bosniak renal cyst classification: 25 years later.Radiology. 2012;262(3):781–785
13. Bosniak MA. The current radiological approach to renal cysts.Radiology. 1986;158(1):1–10
14. Wallis MC, Lorenzo AJ, Farhat WA, Bägli DJ, Khoury AE, Pippi Salle JL. Risk assessment of incidentally detected complex renal cysts in children: potential role for a modification of the Bosniak classification.J Urol. 2008; 180(1):317–321
15. National Cancer Institute at the National Institutes of Health. Wilms tumor and other childhood kidney tumors treatment (PDQ). Available at: www.
cancer.gov/cancertopics/pdq/ treatment/wilms/HealthProfessional/ page2. Accessed March 19, 2013
16. Green DM, Jaffe N. The role of chemotherapy in the treatment of Wilms’tumor.Cancer. 1979;44(1):52–57
17. Gratias EJ, Dome JS. Current and emerging chemotherapy treatment strategies for Wilms tumor in North America.Paediatr Drugs. 2008;10(2): 115–124 [Review]
18. Royer-Pokora B. Genetics of pediatric renal tumors.Pediatr Nephrol. 2013; 28(1):13–23
19. Tagarelli A, Spreafico F, Ferrari A, et al. Primary renal soft tissue sarcoma in children.Urology. 2012;80(3):698–702
20. Castellino SM, Martinez-Borges AR, McLean TW. Pediatric genitourinary tumors.Curr Opin Oncol. 2009;21(3): 278–283
21. Glick RD, Hicks MJ, Nuchtern JG, Wesson DE, Olutoye OO, Cass DL. Renal tumors in infants less than 6 months of age.J Pediatr Surg. 2004;39(4):522–525 [Review]
22. Lowe LH, Isuani BH, Heller RM, et al. Pediatric renal masses: Wilms tumor and beyond.Radiographics. 2000;20(6): 1585–1603 [Review]
23. Fischbach BV, Trout KL, Lewis J, Luis CA, Sika M. WAGR syndrome: a clinical review of 54 cases.Pediatrics. 2005; 116(4):984–988
24. Breslow NE, Norris R, Norkool PA, et al; National Wilms Tumor Study Group. Characteristics and outcomes of children with the Wilms tumor-Aniridia syndrome: a report from the National Wilms Tumor Study Group.J Clin Oncol. 2003;21(24):4579–4585
25. Royer-Pokora B, Beier M, Henzler M, et al. Twenty-four new cases of WT1 germline mutations and review of the literature: genotype/phenotype correlations for Wilms tumor development.Am J Med Genet A. 2004; 127A(3):249–257
26. Pelletier J, Bruening W, Kashtan CE, et al. Germline mutations in the Wilms’ tumor suppressor gene are associated with abnormal urogenital development in Denys-Drash syndrome.Cell. 1991; 67(2):437–447
Tumours (FACT) Collaboration. Constitutional 11p15 abnormalities, including heritable imprinting center mutations, cause nonsyndromic Wilms tumor.Nat Genet. 2008;40(11): 1329–1334
28. Green DM, Breslow NE, Beckwith JB, Norkool P. Screening of children with hemihypertrophy, aniridia, and Beckwith-Wiedemann syndrome in patients with Wilms tumor: a report from the National Wilms Tumor Study. Med Pediatr Oncol. 1993;21(3):188–192
29. DeBaun MR, Siegel MJ, Choyke PL. Nephromegaly in infancy and early childhood: a risk factor for Wilms tumor in Beckwith-Wiedemann syndrome.J Pediatr. 1998;132(3 pt 1): 401–404
30. DeBaun MR, Tucker MA. Risk of cancer during thefirst four years of life in children from The Beckwith-Wiedemann Syndrome Registry.J Pediatr. 1998; 132(3 pt 1):398–400
31. Luithle T, Szavay P, Furtwängler R, Graf N, Fuchs J; SIOP/GPOH Study Group. Treatment of cystic nephroma and cystic partially differentiated nephroblastoma—a report from the SIOP/GPOH study group.J Urol. 2007; 177(1):294–296
32. Ritchey ML, Shamberger RC, Hamilton T, Haase G, Argani P, Peterson S. Fate of bilateral renal lesions missed on preoperative imaging: a report from the National Wilms Tumor Study Group. J Urol. 2005;174(4 pt 2):1519–1521, discussion 1521
33. Lange J, Peterson SM, Takashima JR, et al. Risk factors for end stage renal disease in non-WT1-syndromic Wilms tumor.J Urol. 2011;186(2):378–386
34. Gracia Bouthelier R, Lapunzina P. Follow-up and risk of tumors in overgrowth syndromes.J Pediatr Endocrinol Metab. 2005;18(suppl 1): 1227–1235
35. Lapunzina P. Risk of tumorigenesis in overgrowth syndromes:
a comprehensive review.Am J Med Genet C Semin Med Genet. 2005;137C(1): 53–71
36. Scott RH, Walker L, Olsen ØE, et al Surveillance for Wilms tumour in at-risk children: pragmatic
recommendations for best practice. Arch Dis Child. 2006;91(12):995–999
37. Porteus MH, Narkool P, Neuberg D, et al. Characteristics and outcome of children with Beckwith-Wiedemann syndrome and Wilms’tumor: a report from the National Wilms Tumor Study Group.J Clin Oncol. 2000;18(10): 2026–2031
38. Tan TY, Amor DJ. Tumour surveillance in Beckwith-Wiedemann syndrome and hemihyperplasia: a critical review of the evidence and suggested guidelines for local practice.J Paediatr Child Health. 2006;42(9):486–490
39. Hu M, Zhang GY, Arbuckle S, et al. Prophylactic bilateral nephrectomies in two paediatric patients with missense mutations in the WT1 gene.Nephrol Dial Transplant. 2004;19(1):223–226
40. Rudin C, Pritchard J, Fernando ON, Duffy PG, Trompeter RS. Renal
transplantation in the management of bilateral Wilms’tumour (BWT) and of Denys-Drash syndrome (DDS).Nephrol Dial Transplant. 1998;13(6):1506–1510
41. Aronson DC, Slaar A, Heinen RC, et al Long-term outcome of bilateral Wilms tumors (BWT).Pediatr Blood Cancer. 2011;56(7):1110–1113
42. Hassinger AB, Garimella S. Refractory hypotension after bilateral
nephrectomies in a Denys-Drash patient with phenylketonuria.Pediatr Nephrol. 2013;28(2):345–348
43. Auber F, Jeanpierre C, Denamur E, et al. Management of Wilms tumors in Drash and Frasier syndromes.Pediatr Blood Cancer. 2009;52(1):55–59
44. Schumacher V, Schärer K, Wühl E, et al. Spectrum of early onset nephrotic syndrome associated with WT1 missense mutations.Kidney Int. 1998; 53(6):1594–1600
45. Nso Roca AP, Peña Carrión A, Benito Gutiérrez M, García Meseguer C, García Pose A, Navarro M. Evolutive study of children with diffuse mesangial sclerosis.Pediatr Nephrol. 2009;24(5): 1013–1019
46. Eddy AA, Mauer SM. Pseudohermaphroditism, glomerulopathy, and Wilms tumor (Drash syndrome): frequency in end-stage renal failure.J Pediatr. 1985; 106(4):584–587
47. Argani P, Perlman EJ, Breslow NE, et al. Clear cell sarcoma of the kidney: a review of 351 cases from the National
Wilms Tumor Study Group Pathology Center.Am J Surg Pathol. 2000;24(1):4–18
48. Hiradfar M, Zabolinejad N, Shojaeian R, et al. Pediatric clear cell sarcoma of the kidney with atriocaval thrombus. Pediatr Surg Int. 2012;28(11):1141–1145
49. Gooskens SL, Furtwängler R, Vujanic GM, Dome JS, Graf N, van den Heuvel-Eibrink MM. Clear cell sarcoma of the kidney: a review.Eur J Cancer. 2012; 48(14):2219–2226 [Review]
50. Furtwängler R, Gooskens SL, van Tinteren H, et al. Clear cell sarcomas of the kidney registered on International Society of Pediatric Oncology (SIOP) 93-01 and SIOP 2093-01 protocols: a report of the SIOP Renal Tumour Study Group.Eur J Cancer. 2013;49(16):3497–3506
51. Siegel MJ.Pediatric Body CT. Philadelphia, PA: Lippincott Williams & Wilkins; 1999:226–252
52. Karlsson J, Holmquist Mengelbier L, Ciornei CD, Naranjo A, O’Sullivan MJ, Gisselsson D. Clear cell sarcoma of the kidney demonstrates an embryonic signature indicative of a primitive nephrogenic origin.Genes Chromosomes Cancer. 2014;53(5): 381–391
53. O’Meara E, Stack D, Lee CH, et al. Characterization of the chromosomal translocation t(10;17)(q22;p13) in clear cell sarcoma of kidney.J Pathol. 2012; 227(1):72–80
54. Kalapurakal JA, Perlman EJ, Seibel NL, Ritchey M, Dome JS, Grundy PE. Outcomes of patients with revised stage I clear cell sarcoma of kidney treated in National Wilms Tumor Studies 1-5.Int J Radiat Oncol Biol Phys. 2013;85(2): 428–431
55. Fleitz JM, Wootton-Gorges SL, Wyatt-Ashmead J, et al. Renal cell carcinoma in long-term survivors of advanced stage neuroblastoma in early childhood.Pediatr Radiol. 2003;33(8): 540–545
56. Spreafico F, Collini P, Terenziani M, Marchianò A, Piva L. Renal cell carcinoma in children and adolescents. Expert Rev Anticancer Ther. 2010;10(12): 1967–1978
58. Kumar S, Sharma P, Pratap J, Tiwari P, Bera MK, Kundu AK. Renal cell carcinoma in children and adolescence: our experience.Afr J Paediatr Surg. 2014;11(2):101–104
59. Atilgan D, Uluocak N, Erdemir F, Parlaktas BS, Koseoglu RD, Boztepe O. Multicystic renal cell carcinoma: a rare kidney tumor in children.Urol J. 2013; 10(1):811–814
60. Kesik V, Yalçin B, Akçören Z, Senocak ME, Talim B, Büyükpamukçu M. A rare type of renal cell carcinoma in a girl: hybrid renal cell carcinoma.Pediatr Hematol Oncol. 2010;27(3):228–232
61. Greco AJ, Baluarte JH, Meyers KE, et al. Chromophobe renal cell carcinoma in a pediatric living-related kidney transplant recipient.Am J Kidney Dis. 2005;45(6):e105–e108
62. Said JW, Thomas G, Zisman A. Kidney pathology: current classification of renal cell carcinoma.Curr Urol Rep. 2002;3(1):25–30
63. Jing H, Tai Y, Xu D, Yang F, Geng M. Renal cell carcinoma associated with Xp11.2 translocations, report of a case. Urology. 2010;76(1):156–158
64. Wu A, Kunju LP, Cheng L, Shah RB. Renal cell carcinoma in children and young adults: analysis of clinicopathological, immunohistochemical and molecular characteristics with an emphasis on the spectrum of Xp11.2 translocation-associated and unusual clear cell subtypes.Histopathology. 2008;53(5): 533–544
65. Geller JI, Argani P, Adeniran A, et al. Translocation renal cell carcinoma: lack of negative impact due to lymph node spread.Cancer. 2008;112(7):1607–1616
66. Schafernak KT, Yang XJ, Hsueh W, Leestma JL, Stagl J, Goldman S. Pediatric renal cell carcinoma as second malignancy: reports of two cases and a review of the literature.Can J Urol. 2007;14(6):3739–3744 [Review]
67. Rich BS, McEvoy MP, La Quaglia MP. A case of renal cell carcinoma after successful treatment of Wilms tumor. J Pediatr Surg. 2010;45(9):1883–1886
68. Kubo M, Iwashita K, Oyachi N, Oyama T, Yamamoto T. Two different types of infantile renal cell carcinomas associated with tuberous sclerosis.J Pediatr Surg. 2011;46(10):E37–E41
69. Cohen HT, McGovern FJ. Renal-cell carcinoma.N Engl J Med. 2005;353(23): 2477–2490 [Review]
70. Tsui KH, Shvarts O, Smith RB, Figlin R, de Kernion JB, Belldegrun A. Renal cell carcinoma: prognostic significance of incidentally detected tumors.J Urol. 2000;163(2):426–430
71. Labanaris AP, Schott GE, Zugor V. Renal cell carcinoma in children under 10 years old: a presentation of four cases. Pediatr Surg Int. 2007;23(4):327–330
72. Baek M, Jung JY, Kim JJ, Park KH, Ryu DS. Characteristics and clinical outcomes of renal cell carcinoma in children: a single center experience.Int J Urol. 2010;17(8):737–740
73. Leveridge MJ, Bostrom PJ, Koulouris G, Finelli A, Lawrentschuk N. Imaging renal cell carcinoma with ultrasonography, CT and MRI.Nat Rev Urol. 2010;7(6): 311–325 [Review]
74. Perlman EJ. Pediatric Renal Cell Carcinoma.Surg Pathol Clin. 2010;3(3): 641–651
75. Simpson L, He X, Pins M, et al. Renal medullary carcinoma and ABL gene amplification.J Urol. 2005;173(6): 1883–1888
76. Liu Q, Galli S, Srinivasan R, Linehan WM, Tsokos M, Merino MJ. Renal medullary carcinoma: molecular,
immunohistochemistry, and morphologic correlation.Am J Surg Pathol. 2013;37(3):368–374
77. Pratt CB, Pappo AS. Management of infrequent cancers of childhood. In: Pizzo PA, Poplack DG, eds.Principles and Practice of Pediatric Oncology, 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2004:1158–1160
78. Cook A, Lorenzo AJ, Salle JL, et al. Pediatric renal cell carcinoma: single institution 25-year case series and initial experience with partial nephrectomy.J Urol. 2006;175(4): 1456–1460, discussion 1460
79. Fyfe G, Fisher RI, Rosenberg SA, et al Results of treatment of 255 patients with metastatic renal cell carcinoma who received high-dose recombinant interleukin-2 therapy.J Clin Oncol. 1995; 13(3):688–696
80. Motzer RJ, Mazumdar M, Bacik J, Russo P, Berg WJ, Metz EM. Effect of cytokine therapy on survival for patients with
advanced renal cell carcinoma.J Clin Oncol. 2000;18(9):1928–1935
81. Pizzocaro G, Piva L, Colavita M, et al. Interferon adjuvant to radical nephrectomy in Robson stages II and III renal cell carcinoma: a multicentric randomized study.J Clin Oncol. 2001; 19(2):425–431
82. Escudier B, Michaelson MD, Motzer RJ, et al. Axitinib versus sorafenib in advanced renal cell carcinoma: subanalyses by prior therapy from a randomised phase III trial.Br J Cancer. 2014;110(12):2821–2828
83. Motzer RJ, Escudier B, Bukowski R, et al. Prognostic factors for survival in 1059 patients treated with sunitinib for metastatic renal cell carcinoma.Br J Cancer. 2013;108(12):2470–2477
84. Feldman DR, Baum MS, Ginsberg MS, et al. Phase I trial of bevacizumab plus escalated doses of sunitinib in patients with metastatic renal cell carcinoma.J Clin Oncol. 2009;27(9): 1432–1439
85. Patel PH, Senico PL, Curiel RE, Motzer RJ. Phase I study combining treatment with temsirolimus and sunitinib malate in patients with advanced renal cell carcinoma.Clin Genitourin Cancer. 2009;7(1):24–27
86. Escudier B, Porta C, Bono P, et al. Randomized, controlled, double-blind, cross-over trial assessing treatment preference for pazopanib versus sunitinib in patients with metastatic renal cell carcinoma: PISCES Study. J Clin Oncol. 2014;32(14):1412–1418
87. Molina AM, Hutson TE, Larkin J, et al. A phase 1b clinical trial of the multi-targeted tyrosine kinase inhibitor lenvatinib (E7080) in combination with everolimus for treatment of metastatic renal cell carcinoma (RCC).Cancer Chemother Pharmacol. 2014;73(1): 181–189
88. Glassberg KI. Normal and abnormal development of the kidney: a clinician’s interpretation of current knowledge. J Urol. 2002;167(6):2339–2350, discussion 2350–2351 [Review]
89. Perlman EJ, Faria P, Soares A, et al; National Wilms Tumor Study Group. Hyperplastic perilobar
90. Machmouchi M, Bayoumi M, Mamoun I, Al-Ahmadi K, Kanaan H. Bilateral universal nephroblastomatosis in an 8-month-old infant treated with chemotherapy.Pediatr Nephrol. 2005; 20(7):1007–1010
91. Beckwith JB. Nephrogenic rests and the pathogenesis of Wilms tumor: developmental and clinical
considerations.Am J Med Genet. 1998; 79(4):268–273
92. Tomlinson GE, Breslow NE, Dome J, et al. Rhabdoid tumor of the kidney in the National Wilms’Tumor Study: age at diagnosis as a prognostic factor.J Clin Oncol. 2005;23(30):7641–7645
93. Amar AM, Tomlinson G, Green DM, Breslow NE, de Alarcon PA. Clinical presentation of rhabdoid tumors of the kidney.J Pediatr Hematol Oncol. 2001; 23(2):105–108 [Review]
94. Mehrain R, Nabahati M. A case of rhabdomyosarcoma of kidney mimicking nephroblastoma.Caspian J Intern Med. 2013;4(1):621–623
95. Roberts CW, Biegel JA. The role of SMARCB1/INI1 in development of rhabdoid tumor.Cancer Biol Ther. 2009; 8(5):412–416
96. Versteege I, Sévenet N, Lange J, et al. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer.Nature. 1998;394(6689):203–206
97. Eaton KW, Tooke LS, Wainwright LM, Judkins AR, Biegel JA. Spectrum of SMARCB1/INI1 mutations in familial and sporadic rhabdoid tumors.Pediatr Blood Cancer. 2011;56(1):7–15
98. Biegel JA, Zhou JY, Rorke LB, Stenstrom C, Wainwright LM, Fogelgren B. Germ-line and acquired mutations of INI1 in atypical teratoid and rhabdoid tumors. Cancer Res. 1999;59(1):74–79
99. Radhika S, Bakshi A, Rajwanshi A, et al. Cytopathology of uncommon malignant renal neoplasms in the pediatric age group.Diagn Cytopathol. 2005;32(5): 281–286
100. Ahmed HU, Arya M, Levitt G, Duffy PG, Mushtaq I, Sebire NJ. Part I: Primary malignant non-Wilms’renal tumours in children.Lancet Oncol. 2007;8(8): 730–737 [Review]
101. Ahmed HU, Arya M, Levitt G, Duffy PG, Sebire NJ, Mushtaq I. Part II: Treatment of primary malignant non-Wilms’renal
tumours in children.Lancet Oncol. 2007; 8(9):842–848 [Review]
102. Toutain J, VuPhi Y, Doco-Fenzy M, et al. Identification of a complex 17q rearrangement in a metanephric stromal tumor.Cancer Genet. 2011; 204(6):340–343
103. Chaudry G, Perez-Atayde AR, Ngan BY, Gundogan M, Daneman A. Imaging of congenital mesoblastic nephroma with pathological correlation.Pediatr Radiol. 2009;39(10):1080–1086
104. Irsutti M, Puget C, Baunin C, Duga I, Sarramon MF, Guitard J. Mesoblastic nephroma: prenatal ultrasonographic and MRI features.Pediatr Radiol. 2000; 30(3):147–150
105. Santos LG, Carvalho JS, Reis MA, Sales RL. Cellular congenital mesoblastic nephroma: case report.J Bras Neurol. 2011;33(1):109–112
106. Kelner M, Droullé P, Didier F, Hoeffel JC. The vascular“ring”sign in mesoblastic nephroma: report of two cases.Pediatr Radiol. 2003;33(2):123–128
107. Furtwaengler R, Reinhard H, Leuschner I, et al; Gesellschaft fur Pädiatrische Onkologie und Hämatologie (GPOH) Nephroblastoma Study Group. Mesoblastic nephroma—a report from the Gesellschaft fur Pädiatrische Onkologie und Hämatologie (GPOH). Cancer. 2006;106(10):2275–2283
108. Bayindir P, Guillerman RP, Hicks MJ, Chintagumpala MM. Cellular
mesoblastic nephroma (infantile renal
fibrosarcoma): institutional review of the clinical, diagnostic imaging, and pathologic features of a distinctive neoplasm of infancy.Pediatr Radiol. 2009;39(10):1066–1074
109. Kulkarni MP, Gosavi AV, Anvikar AR, Ramteerthakar NA, Lanjewar DN. Cytodiagnosis of congenital
mesoblastic nephroma: a case report. Diagn Cytopathol. 2013;41(3):234–238
110. Vujanic GM, Sandstedt B, Harms D, Kelsey A, Leuschner I, de Kraker J; SIOP Nephroblastoma Scientific Committee. Revised International Society of Paediatric Oncology (SIOP) working classification of renal tumors of childhood.Med Pediatr Oncol. 2002; 38(2):79–82
111. van den Heuvel-Eibrink MM, Grundy P, Graf N, et al. Characteristics and
survival of 750 children diagnosed with a renal tumor in thefirst seven months of life: A collaborative study by the SIOP/GPOH/SFOP, NWTSG, and UKCCSG Wilms tumor study groups.Pediatr Blood Cancer. 2008;50(6):1130–1134
112. Watanabe N, Omagari D, Yamada T, et al. Anaplastic sarcoma of the kidney: case report and literature review.Pediatr Int. 2013;55(5):e129–e132
113. Gomi K, Hamanoue S, Tanaka M, et al. Anaplastic sarcoma of the kidney with chromosomal abnormality:first report on cytogeneticfindings.Hum Pathol. 2010;41(10):1495–1499
114. Vujanic GM, Kelsey A, Perlman EJ, Sandstedt B, Beckwith JB. Anaplastic sarcoma of the kidney:
a clinicopathologic study of 20 cases of a new entity with polyphenotypic features.Am J Surg Pathol. 2007;31(10): 1459–1468
115. Sebire NJ, Vujanic GM. Paediatric renal tumours: recent developments, new entities and pathological features. Histopathology. 2009;54(5):516–528
116. Wang LL, Perlman EJ, Vujanic GM, et al. Desmoplastic small round cell tumor of the kidney in childhood.Am J Surg Pathol. 2007;31(4):576–584
117. Egloff AM, Lee EY, Dillon JE, Callahan MJ. Desmoplastic small round cell tumor of the kidney in a pediatric patient: sonographic and multiphase CT
findings.AJR Am J Roentgenol. 2005; 185(5):1347–1349
118. Potikyan G, France KA, Carlson MR, Dong J, Nelson SF, Denny CT. Genetically defined EWS/FLI1 model system suggests mesenchymal origin of Ewing’s family tumors.Lab Invest. 2008; 88(12):1291–1302
119. Riggi N, Suvà ML, Suvà D, et al. EWS-FLI-1 expression triggers a Ewing’s sarcoma initiation program in primary human mesenchymal stem cells.Cancer Res. 2008;68(7):2176–2185
120. Tirode F, Laud-Duval K, Prieur A, Delorme B, Charbord P, Delattre O. Mesenchymal stem cell features of Ewing tumors.Cancer Cell. 2007;11(5): 421–429
121. Dela Cruz FS. Cancer stem cells in pediatric sarcomas.Front Oncol. 2013;3:168
Primitive neuroectodermal tumor (PNET)/extraosseous Ewing sarcoma of the kidney.Med Pediatr Oncol. 1998; 30(5):303–307
123. Perer E, Shanberg AM, Matsunaga G, Finklestein JZ. Laparoscopic removal of extraosseous Ewing’s sarcoma of the kidney in a pediatric patient.
J Laparoendosc Adv Surg Tech A. 2006; 16(1):74–76
124. Karpate A, Menon S, Basak R, Yuvaraja TB, Tongaonkar HB, Desai SB. Ewing sarcoma/primitive neuroectodermal tumor of the kidney: clinicopathologic analysis of 34 cases.Ann Diagn Pathol. 2012;16(4):267–274
125. Sausville JE, Hernandez DJ, Argani P, Gearhart JP. Pediatric renal cell carcinoma.J Pediatr Urol. 2009;5(4): 308–314
126. Medeiros LJ, Palmedo G, Krigman HR, Kovacs G, Beckwith JB. Oncocytoid renal cell carcinoma after neuroblastoma: a report of four cases of a distinct clinicopathologic entity.Am J Surg Pathol. 1999;23(7):772–780
127. Wilson CL, Ness KK, Neglia JP, et al. Renal carcinoma after childhood cancer: a report from the childhood cancer survivor study.J Natl Cancer Inst. 2013;105(7):504–508
128. Dash SC, Purohit K, Mohanty SK, Dinda AK. An unusual case of bilateral renal enlargement due to primary renal lymphoma.Indian J Nephrol. 2011;21(1): 56–58
129. Xi S, Chen H, Wu X, et al. Malignant renal angiomyolipoma with metastases in a child.Int J Surg Pathol. 2014;22(2): 160–166
130. Astigueta JC, Abad MA, Pow-Sang MR, et al. Epithelioid angiomyolipoma: a rare variant of renal angiomyolipoma. Arch Esp Urol. 2009;62(6):493–497 [Review]
131. Hilmes MA, Dillman JR, Mody RJ, Strouse PJ. Pediatric renal leukemia: spectrum of CT imagingfindings. Pediatr Radiol. 2008;38(4):424–430
132. Choh NA, Choh SA, Yousuf R, Jehangir M. A giant aneurysm arising from renal angiomyolipoma in tuberous sclerosis. Arch Dis Child. 2009;94(1):47–48
133. Khaled A, Arif N, Zaman W, Nasir TA. Bilateral renal angiomyolipoma not associated with tuberous sclerosis:
a case report. Available at: www. banglajol.info/index.php/PULSE/article/ download/6965/529. Accessed May 30, 2014
134. Ito M, Sugamura Y, Ikari H, Sekine I. Angiomyolipoma of the lung.Arch Pathol Lab Med. 1998;122(11): 1023–1025
135. Umeoka S, Koyama T, Miki Y, Akai M, Tsutsui K, Togashi K. Pictorial review of tuberous sclerosis in various organs. Radiographics. 2008;28(7):e32
136. Milner J, McNeil B, Alioto J, et al. Fat poor renal angiomyolipoma: patient, computerized tomography and histologicalfindings.J Urol. 2006; 176(3):905–909
137. Kim JK, Park SY, Shon JH, Cho KS. Angiomyolipoma with minimal fat: differentiation from renal cell carcinoma at biphasic helical CT. Radiology. 2004;230(3):677–684
138. Seyam RM, Bissada NK, Kattan SA, et al. Changing trends in presentation, diagnosis and management of renal angiomyolipoma: comparison of sporadic and tuberous sclerosis complex-associated forms.Urology. 2008;72(5):1077–1082
139. Inci O, Kaplan M, Yalcin O, Atakan IH, Kubat H. Renal angiomyolipoma with malignant transformation,
simultaneous occurrence with malignity and other complex clinical situations.Int Urol Nephrol. 2006;38(3-4):417–426
140. Kohashi K, Oda Y, Nakamori M, et al. Multifocal metanephric adenoma in childhood.Pathol Int. 2009;59(1):49–52
141. Mei H, Zheng L, Zhou C, Tong Q. Metanephric adenoma in a 2-year-old child: case report and
immunohistochemical observations. J Pediatr Hematol Oncol. 2010;32(6): 489–493
142. Küpeli S, Baydar DE, Canakl F, et al. Metanephric adenoma in a 6-year-old child with hemihypertrophy.J Pediatr Hematol Oncol. 2009;31(6):453–455
143. Davis CJ Jr, Barton JH, Sesterhenn IA, MostofiFK. Metanephric adenoma. Clinicopathological study offifty patients.Am J Surg Pathol. 1995;19(10): 1101–1114
144. Jones EC, Pins M, Dickersin GR, Young RH. Metanephric adenoma of the
kidney. A clinicopathological,
immunohistochemical,flow cytometric, cytogenetic, and electron microscopic study of seven cases.Am J Surg Pathol. 1995;19(6):615–626
145. McNeil JC, Corbett ST, Kuruvilla S, Jones EA. Metanephric adenoma in afi ve-year-old boy presenting with chyluria: case report and review of literature.Urology. 2008;72(3):545–547
146. Liniger B, Wolf RW, Fleischmann A, Kluwe W. Local resection of metanephric adenoma with kidney preservation.J Pediatr Surg. 2009; 44(8):E21–E23
147. Navarro O, Conolly B, Taylor G, Bägli DJ. Metanephric adenoma of the kidney: a case report.Pediatr Radiol. 1999; 29(2):100–103
148. Pins MR, Jones EC, Martul EV, Kamat BR, Umlas J, Renshaw AA. Metanephric adenoma-like tumors of the kidney: report of 3 malignancies with emphasis on discriminating features.Arch Pathol Lab Med. 1999;123(5):415–420
149. Renshaw AA, Freyer DR, Hammers YA. Metastatic metanephric adenoma in a child.Am J Surg Pathol. 2000;24(4): 570–574
150. Drut R, Drut RM, Ortolani C. Metastatic metanephric adenoma with foci of papillary carcinoma in a child: a combined histologic,
immunohistochemical, and FISH study. Int J Surg Pathol. 2001;9(3):241–247
151. Nakagawa T, Kanai Y, Fujimoto H, et al. Malignant mixed epithelial and stromal tumours of the kidney: a report of the
first two cases with a fatal clinical outcome.Histopathology. 2004;44(3): 302–304
152. Mehra BR, Thawait AP, Akther MJ, Narang RR. Multicystic nephroma masquerading as Wilms’tumor: a clinical diagnostic challenge.Saudi J Kidney Dis Transpl. 2011;22(4):774–778
153. Babu S, Agarwal R, Narayansamy K, Rajendranath R, Balasubramanian S. Cystic renal neoplasm causing hypertension in a 2-year-old child. Saudi J Kidney Dis Transpl. 2011;22(4): 779–781
155. Kacar A, Azili MN, Cihan BS, Demir HA, Tiryaki HT, Argani P. Metanephric stromal tumor: a challenging diagnostic entity in children.J Pediatr Surg. 2011;46(12):e7–e10
156. Argani P, Beckwith JB. Metanephric stromal tumor: report of 31 cases of a distinctive pediatric renal
neoplasm.Am J Surg Pathol. 2000; 24(7):917–926
157. Argani P. Metanephric neoplasms: the hyperdifferentiated, benign end of the Wilms tumor spectrum?Clin Lab Med. 2005;25(2):379–392 [Review]
158. De Pasquale MD, Diomedi-Camassei F, Serra A, et al. Recurrent metanephric stromal tumor in an infant.Urology. 2011;78(6):1411–1413
159. Hu J, Wu Y, Qi J, Zhang C, Lv F. Ossifying renal tumor of infancy (ORTI): a case report and review of the literature. J Pediatr Surg. 2013;48(2):e37–e40
160. Liu J, Guzman MA, Pawel BR, et al. Clonal trisomy 4 cells detected in the ossifying renal tumor of infancy: study of 3 cases.Mod Pathol. 2013;26(2): 275–281
161. Vazquez JL, Barnewolt CE, Shamberger RC, Chung T, Perez-Atayde AR. Ossifying renal tumor of infancy presenting as a palpable abdominal mass.Pediatr Radiol. 1998;28(6):454–457
162. Xu B, Zhang Q, Jin J. Hypertension secondary to reninoma treated with laparoscopic nephron-sparing surgery in a child.Urology. 2012;80(1):210–213
163. Gottardo F, Cesari M, Morra A, Gardiman M, Fassina A, Dal Bianco M. A kidney tumor in an adolescent with severe hypertension and hypokalemia: an uncommon case—case report and review of the literature on reninoma. Urol Int. 2010;85(1):121–124
164. Dong D, Li H, Yan W, Xu W. Juxtaglomerular cell tumor of the kidney—a new classification scheme. Urol Oncol. 2010;28(1):34–38
165. Markey RB, MacLennan GT. Juxtaglomerular cell tumor of the kidney.J Urol. 2006;175(2):730
166. Duan X, Bruneval P, Hammadeh R, et al. Metastatic juxtaglomerular cell tumor in a 52-year-old man.Am J Surg Pathol. 2004;28(8):1098–1102
MUSICFORMUSCLES:On most mornings or evenings during the winter, I exercise in
our basement. I may run on the treadmill, ride on a stationary bike trainer, or lift weights. Since I do not usually have much spare time, I tend to do interval training in which I exercise as hard as I can for short periods of time, followed by rest or exercise at a much lower intensity. The one constant to my routine is that I always play music at high volume. My kids tease me about my playlists, but I alwaysfind the workouts less tedious or painful when I listen to music. While I am a bit concerned that I may not be doing my ears any favors, new data suggest that music can indeed improve high intensity exercise performance.
As reported inThe New York Times(Well: October 22, 2014), scientists recruited 20 young adults and introduced them to interval training on a stationary bike. They measured pedaling power output and asked each to rate how hard the exercise felt. The researchers then built custom playlists for each participant based on individual preferences. Participants then participated in two more interval training exercises– one while listening to music and the other without. Participants graded each session as similarly very hard. However, pedaling power output was much higher in the session with music. In other words, music did not change the perception of the workout, but did improve performance. All riders said that if they were to take up interval training, they would include music.
While I do not measure my performance in the basement, I do know that the blaring, upbeat music seems to help. Now that I have some data to back me up, maybe my kids will not laugh quite so hard at me.