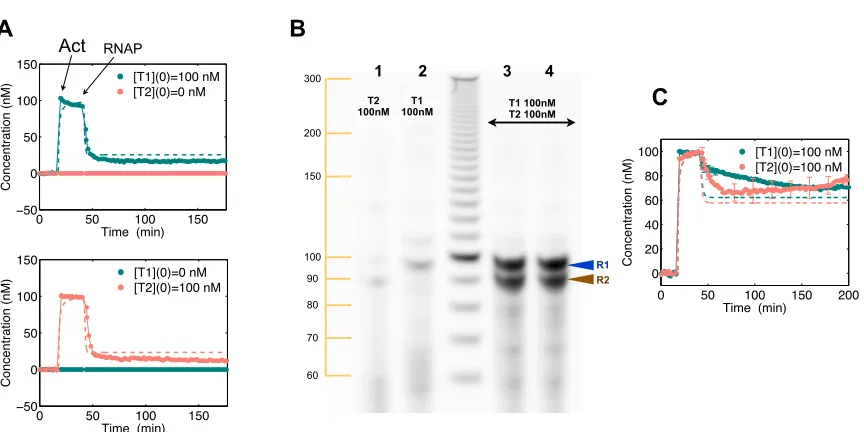
Analysis, design, and
in vitro
implementation of robust
biochemical networks
Thesis by
Elisa Franco
In Partial Fulfillment of the Requirements
for the Degree of
Doctor of Philosophy
California Institute of Technology
Pasadena, California
2012
c
2012
Elisa Franco
Acknowledgements
My years at Caltech have been a true learning journey: I am grateful to many, many people for
accompanying me through it.
First of all, I want to thank my advisor Richard Murray, who made the journey possible and
provided me with invaluable guidance. His enthusiasm, support, and vision turned into exciting
chal-lenges many of what I thought were research dead ends or unsurmountable issues. I am very grateful
to Erik Winfree, who generously allowed me to become an extended member of his laboratory. His
sharp ideas, constructive criticism, and amazing attention to detail have been essential stepping
stones for the development of my graduate work. I would also like to thank Paul Rothemund and
John Doyle for being part of my thesis committee.
My deepest gratitude goes to Jongmin Kim, for introducing me toin vitro synthetic circuits and
being a wonderful collaborator and friend for many years. I am also thankful to all the members of
the Winfree lab, and in particular to Nadine Dabby, Rebecca Schulman, and Rizal Hariadi for their
support, feedback, and friendship.
I am in great debt to my friend Franco Blanchini: he helped turn a casual conference hallway
conversation into a good research project. I also want to thank Domitilla Del Vecchio for helpful
discussions and advice. I am grateful for having collaborated with John Dabiri at the beginning of
my graduate studies.
I was lucky to work with bright and curious undergraduates: Per-Ola Forsberg, with whom I
started the flux regulation project, Chris Sturk, and Fei Chen.
All the members of the Murray group have been extraordinary journey companions – literally so,
during the fun and adventurous Murray group trips. In particular, I want to thank Mary Dunlop,
Julia Braman, Vanessa J¨onsson, Ophelia Venturelli, Tim Chung, Pete Trautman, Marcos Nahmad,
Dionysios Barmpoutis, and Shuo Han. I am also in debt to Dennice Gayme and Genti Buzi for
our “job search” sessions, and to Katie Galloway and Josh Michener for the stimulating bio-control
journal club meetings.
For helping with laboratory supplies, traveling, and many other the practical details of my
doctorate years, I am grateful to Gloria Bain, Karolyn Knoll, and Anissa Scott.
harder process: in particular, I would like to thank Daniela Maniezzo, Maria Rita D’Orsogna,
Guido Maretto, Roberto Congiu, Martina Carbone, Diego Fazi, Pierpaolo Bergamo, Luca Giacchino,
Riccardo Schmid, Andrea Censi, and Luigi Perotti.
Finally, I have to thank my family for their endless support. In particular, my mother Marina,
my aunt Lucia, and my husband Mohsen, for their love and patience; and my old friend Tariq for
Abstract
The functionalities of every living organism are wired in the biochemical interactions among
pro-teins, nucleic acids, and all the other molecules that constitute life’s building blocks. Understanding
the general design principles of this “hardware of life” is an exciting and challenging task for modern
bioengineers. In this thesis, I focus on the topic of molecular network robustness: I investigate
several design rules guaranteeing desired functionalities in specific systems, despite their
compo-nents variability. Experimental verifications of such design schemes are carried out using in vitro
transcriptional circuits, a minimal analogue of cellular genetic networks.
The first problem I consider is flux control, which is a fundamental feature for the correct
performance of biochemical systems. I describe a simple model problem where two reagents bind
stoichiometrically to form an output product. In the absence of any regulation, imbalances in the
reagent production rates can cause accumulation of unused molecules, and limit the output flow. To
match the reagents’ flux robustly with respect to the open loop rates, I propose the use of negative
or positive feedback schemes that rely on competitive binding. Such schemes are modeled through
ordinary differential equations and implemented using transcriptional circuits; data are presented
showing the performance of the two approaches.
The second topic I examine is the functional robustness of interconnected networks. Molecular
devices characterized in isolation may lose their properties once interconnected. This challenge
is illustrated with a case study: a synthetic transcriptional clock is used to time conformational
changes in a molecular nanomachine called DNA tweezers. Mass conservation introduces parasitic
interactions that perturb the oscillator trajectories proportionally to the total amount of tweezers
“load”. To overcome this problem, we can use a transcriptional switch that acts as a buffer amplifier,
achieving signal propagation and at the same time reducing the perturbations on the source of signal.
Finally, I describe a general class of control-theoretic methods to analyze structural robustness
in natural biological systems. Using Lyapunov theory and set invariance, the stability properties
of several well-known case studies are analytically demonstrated. The key feature of this analysis
is its reliance on parameter-independent models, which only capture essential dynamic interactions
Contents
Acknowledgements iv
Abstract vi
1 Introduction 1
1.1 Design principles for robust molecular networks . . . 1
1.2 Cell-free methods . . . 3
1.2.1 In vitro transcriptional circuits . . . 4
1.3 Thesis overview and contribution . . . 8
2 Flux control for biochemical networks 11 2.1 Introduction . . . 11
2.2 Problem formulation . . . 12
2.2.1 Self-repression . . . 13
2.2.2 Cross-activation . . . 17
2.3 Implementation with transcriptional circuits . . . 21
2.3.1 Self-repression . . . 21
2.3.1.1 Modeling . . . 22
2.3.1.2 Experimental results . . . 25
2.3.1.3 Materials and methods . . . 30
2.3.2 Cross-activation . . . 32
2.3.2.1 Modeling . . . 32
2.3.2.2 Preliminary experimental results . . . 37
2.3.2.3 Materials and methods . . . 38
2.4 Discussion . . . 42
3.2 Problem formulation . . . 45
3.3 Experimental results . . . 49
3.3.1 Synthesis of a molecular oscillator using transcriptional circuits . . . 49
3.3.2 A simple load mechanism: molecular tweezers . . . 51
3.3.3 Coupling the oscillator to the tweezers load: signal transmission and back-action 52 3.3.4 Implementing an insulation component . . . 56
3.4 Modeling . . . 58
3.5 Discussion . . . 60
3.6 Materials and methods . . . 62
3.7 Appendix . . . 65
3.7.1 Simple model for the oscillator: load coupling and insulation . . . 65
3.7.1.1 A simple model for the transcriptional oscillator and its non-dimensional version . . . 65
3.7.1.2 Oscillator coupled to a molecular load and stationary approximation 68 3.7.1.3 Insulation . . . 78
3.7.2 Relevant sequence interactions . . . 83
3.7.3 Sample notation . . . 93
3.7.4 Sample preparation . . . 93
3.7.5 Fluorescence data processing . . . 95
3.7.6 T12-channel data . . . 98
3.7.7 Analysis of the oscillations . . . 100
3.7.8 Day-to-day variability . . . 101
3.7.9 Set-to-set variability . . . 102
3.7.10 Oscillation period . . . 104
3.7.11 Oscillation amplitude . . . 105
3.7.12 Effects of the load on the oscillator performance . . . 105
3.7.13 Leak transcription from off-state switches . . . 110
3.7.14 Lack of transcription from T21·rA1 complex . . . 111
3.7.15 Interactions between enzymes and tweezers . . . 112
3.7.16 Effects of changing enzyme volume ratio . . . 114
3.7.17 Effects of changing the DNA thresholds . . . 115
3.7.18 Overview of all fluorescence data sets collected at Caltech . . . 116
4 Structural robustness in molecular networks 121 4.1 Introduction . . . 121
4.3 Results and discussion . . . 128
4.3.1 The L-arabinose network . . . 129
4.3.2 The sRNA pathway . . . 130
4.3.3 The cAMP dependent pathway . . . 136
4.3.4 Thelac operon . . . 138
4.3.5 The MAPK signaling pathway . . . 140
4.4 Conclusions . . . 143
5 Summary and future work 145 5.1 Flux regulation . . . 145
5.2 Oscillatory systems . . . 146
5.3 Robustness in molecular networks . . . 147
Chapter 1
Introduction
1.1
Design principles for robust molecular networks
All living organisms, from bacteria to humans, share a remarkable feature: to survive, they must
be able to sense external stimuli and implement adequate responses. The ability to effectively
control their own behavior based on the “measured” environment is what makes individuals fit and
successful. But how do living things make decisions that are crucial to their survival? This question
branches out in many directions: from neuro-economics to ethology to molecular biology, several
research fields have focused on different aspects of how “control” happens at every layer of what we
call life.
At the simplest level, we find that single cells are individually capable of interacting with their
surroundings: in this context, decision making and control are embedded in biochemical events. One
of the most classical examples is given by the famous experiments of Jacob and Monod [53] in the
1960s, which showed thatE. coli adapts its gene expression profile to the type of nutrient available.
When lactose is abundant, but glucose is not, a set of genes called the lac operon is activated
through a lactose-dependent cascade of reactions. The proteins expressed from thelacoperon allow
the cells to metabolize lactose and grow. In the absence of lactose, or when glucose is present at
high concentrations, thelac operon genes are repressed: thus, cells do not waste energy to produce
unnecessary lactose-digesting enzymes. This is a clear example of how the control center of a cell is
in large part constituted by chemical reactions. It is appropriate to classify a set of molecules that
interact and thereby induce specific cellular behaviors as a molecular or biochemical network.
Continuing with our example, the metabolites, genes, and proteins involved in the lac operon
genetic switch should respond consistently to variations in the available nutrient. However, cells
generally differ from each other in size, and therefore in the number and distribution of metabolites
and proteins present. Moreover, the intracellular environment is crowded and its content is affected
by several parameters, such as temperature and external inputs. Potentially, undesired interactions
network behavior.
So are there features that confer robustness to a biochemical network? Although evolution
op-erates more as a tinkerer than as an engineer [6], several examples of engineering design principles
have been identified in biological systems. For example, negative feedback is the control theorists’
favorite tool to confer robustness to a system [12], because it structurally reduces the impact of
parametric uncertainty and disturbances. In the biological world, there is evidence that negative
auto-regulation in gene expression reduces the variability of protein concentration in cellular
popu-lations [16]. Negative feedback has also been related to the response robustness (and speed) of the
heat shock response inE. coli [31].
A classical example of robust molecular circuitry is probably given by bacterial chemotaxis [14,
9, 117]. The action of the flagellar motor ofE. coli is driven by a cascade of signaling proteins, whose
active or inactive state is determined by the presence of nutrient in the environment. Both analysis
on a simplified ordinary differential equation (ODE) model [14] and experiments [9] showed how the
E. coli flagellar motion presents a robustly stable steady-state: steps in the nutrient concentration
only temporarily alter the motor equilibrium. Cells are therefore sensitive to nutrient gradients,
but always return to their steady state motion (such property is also referred to as adaptability).
Such stable steady state can be described as a function of the concentrations of the signaling
cas-cade protein components and a few binding rates, and is therefore independent of external inputs.
Further analysis also demonstrated how integral feedback is present in the chemotaxis network, and
guarantees robustness (perfect adaptation) of the equilibrium [134].
Experimental and theoretical studies aimed at unraveling the design principles of existing
biologi-cal networks generally fall under the category of systems biology. A different approach is represented
by synthetic biology [87], which instead focuses on the design of new biological circuits. However,
creating new functionalities can be also useful for probing existing systems. On the one hand, for
instance, bacteria and yeast have been engineered to become micro-scale factories to produce fuel,
anti-malarial drug precursors, insulin, and even silk [69, 100, 131]. On the other hand, we can cite
the example of the MAPK pathway synthetic re-wiring, which has been extremely helpful in
clari-fying the role of several proteins involved in the cascade [17, 15]. Another class of examples is given
by the many artificial oscillators synthesized in the past decade [13, 25, 33, 44, 128], which provide
insights into the design principles underlying natural cellular clocks and circadian rhythms.
Robust-ness of negative-feedback-loop-based oscillators, for instance, has been experimentally linked to the
presence of delays [122], in agreement with classical control theory results [99, 11]. The synthetic
approach has also given interesting insights regarding organism-level network robustness: in [52],
for instance, it was demonstrated that survival ofE. coli was not significantly altered by promoter
recombinations adding new links across different networks. Some of the re-wired networks actually
It is imperative to characterize and study molecular networks in their own operational context,
the cell. However, the complexity of the cellular environment may be an insurmountable obstacle to
a detailed understanding of molecular interactions. In fact, quantitative predictions on the dynamic
behaviors of in vivo molecular networks are limited to small systems, mostly due to the lack of
knowledge of the system parameters and to the presence of unmodeled reactions. Synthetic, cell-free
biochemical approaches offer a bottom-up, simplified alternative to the study of molecular circuitry.
1.2
Cell-free methods
Operating in an in vitro environment with a limited number of biological parts offers several
ad-vantages. First, many layers of complexity present in vivo may be eliminated, allowing scientists
to focus on specific phenomena more quantitatively. Second, fully artificial biological design
princi-ples and chemistries can be explored, opening new doors for technology and for understanding the
evolution of life.
Cell-free transcription and translation regulatory circuits have been successfully reproduced
in [90], with the purpose of achieving a high level of detail (relative to in vivo studies) in the
investigation of genetic network behaviors. A good example of howin vitro assays can reveal new
information about natural networks is given by [88], where the reconstruction of circadian
oscilla-tions of cyanobacterial KaiC phosphorylation showed that this process is independent of
transcrip-tion and translatranscrip-tion. Recently, a similar in vitro set of experiments showed that the dynamics of
this oscillator are determined by intermolecular associations: for instance, mutations altering the
binding rates of KaiB to KaiC will modulate the oscillator period [96]. A faithful reproduction
of in vitro cellular environment is still challenging, requiring many components [112] or
not-well-characterized extracts [90]. However, transcription-translation kits for cell-free protein production
are now commercially available; such kits are particularly useful for the synthesis of unnaturally
modified aminoacids [114, 113].
The quest for the minimal biochemistry that supports life [125, 73] is another area wherein vitro
experiments are essential. A related topic of great interest is the role of nucleic acids in general, and
of RNA in particular, in the development of life and regulation of gene expression [38, 22].
In vitro synthetic biology and nanotechnology are rapidly evolving [119, 30] in many directions:
one relevant trend is the use of nucleic acids for the implementation of natural algorithms and
chemical reaction networks. The most attractive property of nucleic acids is programmability [118]:
established methodologies are available to reliably predict structure and hybridization pathways of
an oligonucleotide molecule, starting from its plain sequence information [141, 80, 2, 28]. If we
can predict the structure of a given nucleic acid strand, the ability to design systems of strands
nucleic acids have been designed to self-assemble into arbitrary shapes [102, 59]; to create devices
moving on programmed paths [74, 137] and performing tasks [47]; and to construct biochemical logic
circuitry [110, 140] and molecular machines [139, 24].
The programmability of nucleic acids makes them an ideal candidate for theoretical and
experi-mental studies regarding general chemical reaction networks. In [120], the authors propose motifs for
the implementation of arbitrary chemical dynamics with nucleic acids: such dynamics are generated
through toehold-mediated branch migration [138, 111], and their speed can be tuned by suitably
choosing the length of the toehold domains. (I will return to the topic of branch migration in
Sec-tion 1.2.1.) Numerical tools for the automated generaSec-tion of DNA strands implementing a desired
reaction network are also available [93].
Although nucleic acid catalytic devices are available, it is interesting to explore the computational
and dynamical capabilities of systems integrating proteins and nucleic acids. This is an attractive
setup for two main reasons: first, we have a chance to work with molecular network scenarios that
may be closer to those of natural networks; second, we can develop useful ground knowledge for
the simultaneous programmability of both nucleic acids and amino-acid sequences. Predicting and
programming enzyme folding and function is a very active research area [77]: however, custom
protein synthesis (with a specified structure and function) is still not possible.
One of the first attempts to constructin vitromolecular circuitry using DNA and proteins is the
predator-prey system in [4], which consisted of DNA templates and only three proteins: T7 RNA
polymerase, M-MLV reverse transcriptase, and RNase H cloned from E. coli. The accumulation
of sequence mutations is one of the likely reasons for the limited success of those experiments.
More recently, logic gates using several enzymes [126] and full metabolic platforms [55] have been
characterized. Transcriptional circuits, developed by J. Kim in the Winfree lab at Caltech, are a
versatile tool for building molecular networks, and will be described in detail in the next section.
1.2.1
In vitro
transcriptional circuits
Synthetic in vitro genetic transcriptional circuits [61, 63] consist of nucleic acids and two protein
species, T7 RNA polymerase (RNAP) and E. coli RNase H. Here I will describe their general
features, providing the relevant background information for Chapters 2 and 3 of this thesis. Starting
with Figure 1.1 A, from now on nucleic acids will be graphically represented as linear strings of
letters corresponding to their bases (the helical geometry of double-stranded DNA and RNA will
not be shown); the backbone 5’-3’ direction will be indicated with an arrow at the 3’ end. When
appropriate, specific functional areas, or domains, of a nucleic acid strand will be associated with
different colors (e.g., domains d1 and d2 in Figure 1.1 A); complementary strands will have the same
color (e.g., domains d1 and d1’ in Figure 1.1 A).
A
B
C
D
TGAACGAACGACACTAATG
!
AACTAC
GTAGTTCATTAGTGTCGTTCGTTCA
!
GTGTAGTGTTG
d1 d2
d1’
E
ON OFF + activator
T7 promoter
+
+
k+
k-35 bases
C=A’
C=A’ A
B
35 bases
25 bases
A B
∆G -60kcal/mol
∆G -40kcal/mol X
Y Z=X’
Z=X’ X
Y
Terminator
toehold 4-10 bases Consensus domain 17-20 bases
T7 promoter, 17 bases
Transcribed domain
Functional domain
Activator
OFF ON
ACTIVATOR
INHIBITOR
TRANSCRIPTION Rp
Rh (if RNA inhibitor)
-12
+1
A
I
T T•A
O
A A•I
A
Template strand Non-template strand
Input domain
5’ 3’
30 bases
[image:14.612.150.498.81.502.2]-50kcal/mol
one (or more) inputs and generating one (or more) outputs, which can be used to interconnect
different switches [61]. Such switches can be implemented as short, linear, synthetic genes whose
activity can be turned on and off by altering their promoter region. From now on I will refer to
these short artificial genes in transcriptional networks as templates or “genelets”, a term originally
suggested by Prof. E. Klavins. I will now introduce two notions that are helpful for understanding
how the state of such genelets can be systematically switched.
•Switching promoter activity: Promoters are double-stranded genetic domains having a high
binding affinity for RNA polymerase. The binding affinity can be lost when the structure [57] or
sequence [49] of the promoter region is altered, resulting in weaker transcription of the downstream
region. A promoter that is partially single-stranded, where the template strand is missing, does
not represent a good binding site for RNAP [57]. Referring to Figure 1.1 B, top, if the non-coding
strand of the promoter is single-stranded, the genelet can be effectively considered off. The
tran-scription rate of this incomplete promoter is, in general, below 10% of the trantran-scription rate of
a fully double-stranded promoter. This residual transcription activity is here called transcription
“leak”, and we find that it is dependent on the promoter flanking sequences 3.7.13. When a DNA
strand complementary to the promoter single-stranded domain is added in solution, the
transcrip-tion efficiency is recovered and the gene can be considered on. (Data comparing the on and off
transcription efficiency of some of the genelets used in this thesis are shown in Section 3.35.) The
single-stranded DNA species switching on the genelet will be called an activator. Details regarding
the optimal design of the nicked promoter can be found in [61], Section 3.4. So far, only the
bacte-riophage T7 promoter has been used in transcriptional circuits, due to its high binding affinity and
transcription efficiency for the T7 RNA polymerase enzyme, which is commercially available from
most biotechnology vendors.
•Branch migration: Consider the two nucleic acid complexes shown in Figure 1.1 C, top. One is formed by strands X and Y, the second is a single-stranded species Z, which is fully complementary
to X. The complex formed by strands X and Y is partially single-stranded: the blue overhang
is an exposed domain, to which the corresponding blue domain of strand Z will initiate binding,
subsequently peeling off X from Y. In fact, the system switches quickly to a final, thermodynamically
more favorable configuration, where X is bound to its complement Z=X’ and Y is released in solution.
The blue overhang, where the migration of strands is initiated, is called a toehold. The speed of
the reaction is determined by the length of the toehold, as shown in [138] through fluorescence
experiments.
The two above notions can be combined: a genelet may be designed to be switched on by
an activator strand added in solution, and switched off by branch migration. Branch migration
complex, stripping off the activator strand. General genelet design specifications for the required
domains and their lengths, are shown in Figure 1.1 D. The overall mechanism for switching on and
off a genelet is depicted in Figure 1.1 E.
Genelets can be interconnected through their RNA outputs by means of an inhibition or
acti-vation pathway. The RNA output of a genelet can serve as an inhibitor for a downstream genelet;
alternatively, the RNA output can be used to release an activator otherwise sequestered in an
ac-tivator/inhibitor complex. RNA has the potential to activate a DNA template by binding to the
single-stranded activation domain, thereby completing the promoter; however, due to the constraints
of our system, this is pathway is not used [82]. Degradation is introduced in the system using the
endonuclease RNase H, which targets DNA-RNA hybrids, hydrolyzing the RNA strand and releasing
the DNA strand.
The general theoretical foundations for transcriptional circuits were laid out in [62], where the
computational capability of these molecular networks is demonstrated to be equivalent to that
of neural networks. In general, it is possible to systematically model these circuits using ODEs.
(Typically, transcriptional circuits experiments are run at high molecular counts: stochasticity can
be safely neglected.) For instance, referring to Figure 1.1 E, consider a genelet T having a DNA
activator A, an RNA inhibitor I, and an RNA output O. The chemical reactions expected to occur
by design are:
Activation T + AkTA
→ T·A
Inhibition T·A + IkTAI→ T + I·A
Annihilation A + IkAI
→A·I
Transcription: on state RNAP + T·A
k+ON → ← k−ON
RNAP·T·AkcatON
→ RNAP + T·A + O (1.1)
Transcription: off state RNAP + T
k+OFF → ← k−OFF
RNAP·TkcatOFF
→ RNAP + T + O
Degradation RNaseH + A·I
k+H → ← k−H
RNaseH·A·IkcatH
→ RNaseH + A.
(All hybridization reactions are reversible, but the reverse reaction is extremely slow and can be
neglected in practice.) The corresponding ODEs can be derived immediately, following the general
rules for mass action kinetics. In general, nucleic acid hybridization rates can be measured or
estimated from the literature, while enzymatic parameters are more difficult to establish and have a
higher variability [61, 63, 64]. (Enzymatic parameter uncertainty will be discussed in particular in
The concentrations of activators and inhibitors represent tunable thresholds. Branch
migra-tion reacmigra-tions yielding inhibimigra-tion, annihilamigra-tion, or activamigra-tion are stoichiometric, competitive binding
processes. Competitive binding easily generates ultrasensitive responses of the switches [21, 81]:
this is an important design feature of transcriptional circuits, and is particularly crucial to achieve
oscillatory dynamics.
Several networks have been experimentally characterized using transcriptional circuits:
self-inhibiting and self-activating genelets [61], a bistable toggle switch [63], and negative-feedback-based
oscillators differing for their topology [64]. In this thesis, I will use this tool kit to construct systems
achieving robust properties to be defined later.
1.3
Thesis overview and contribution
Let us go back to our initial question: what are the features that confer robustness to a biochemical
network? In this thesis, I will focus on three different topics related to this question. Two chapters
include work that follows a “synthetic”, bottom-up approach: I will consider specific robust design
objectives for biochemical networks, followed by synthesis using transcriptional circuits. The last
chapter will instead follow a “systems” approach, reporting more general theoretical robustness
results for existing molecular pathways.
•Chapter 2: Flux control for molecular networks. Flux control is a fundamental feature
for the correct performance of large scale networks, of which familiar examples are the Internet,
power grids, or even pipe networks. In the biological world, cells rely as heavily for their survival
on a regulated flow of nucleic acids, transcription factors, and other metabolites. It is therefore
interesting to explore and understand molecular flow rate control at the molecular level, especially
to develop systematic design principles for large biochemical circuits.
In this chapter I will propose two network architectures based on negative and positive feedback, to
regulate and match the output flow rate of two interconnected systems. Feedback is implemented
through mass action chemical reactions, which down- or up-regulate the activity of the molecules
generating the network output. To my knowledge, this design has not been considered elsewhere
in the literature. First, negative auto-regulation and positive cross-regulation will be introduced
through a very simple, intuitive ODE model. Then, I will describe the implementation of these
networks using transcriptional circuits, showing preliminary experimental results. Numerical
sim-ulations and data suggest that feedback confers robustness to the system with respect to certain
parametric variations and to initial conditions.
The general idea of flux control through positive and negative feedback has been previously presented
the negative auto-regulation circuit; the first experiments and numerical simulations were carried
out by an undergraduate student, Per-Ola Forsberg (SURF program at Caltech). Richard Murray
suggested studying the cross-activation scheme. All the analysis, data, and numerical simulations
reported in this thesis were performed by me.
•Chapter 3: Modularity of interconnected systems. An important research direction in
synthetic biology is the systematic design and construction of large molecular networks. Ideally,
biological devices should behave modularly, i.e., they should maintain their functionalities
(charac-terized in isolation) when interconnected to other devices. This can be rephrased as a question of
robustness: by design, the properties of a system should not be disrupted by the interconnection with
other systems. Achieving modularity is a challenge in most engineering fields: classical examples
include voltage drops at the output of non-ideal voltage generators, pressure losses in pipe networks
and level changes in systems of tanks.
This chapter is dedicated to the experimental study of a molecular oscillator to be used as a clock for
a downstream molecular device. Mathematical modeling and experiments show that interconnecting
the oscillator to its load in a direct manner, i.e., by stoichiometric binding and release, results in
undesired back-action effects and loss of the original signal. Loosely speaking, the back-action is
primarily caused by mass conservation constraints. This issue is mitigated by the introduction of a
molecular insulator, a node draining a small amount of molecules from the oscillator and using them
to amplify its signal [27]. Experiments are carried out using the tool kit of transcriptional circuits.
The project presented in this chapter was developed in close collaboration with the group of Prof.
Friedrich Simmel at the Technical University in Munich. F. Simmel and E. Friedrichs had the original
idea of using the transcriptional oscillator proposed in [64] to time conformational switching in the
well-known molecular tweezers system [139]. Jongmin Kim initially suggested connecting another
genelet to the oscillator, using its RNA output to induce switching in the tweezers; this eventually
became our insulator design. My contribution was the idea of using this system as a benchmark to
study the general challenges of molecular modularity and insulation; such idea was largely inspired
by [27] and by several discussions with Prof. Domitilla Del Vecchio. While several experiments I
performed were originally designed by the group at TUM, I developed many control experiments to
better understand the retroactivity effects and the tweezers behavior. Specific challenges I tackled
were data reproducibility, oscillation frequency and amplitude tuning, and the development of a
new transcription protocol to avoid the use of commercial kits. In this thesis I will only report
experiments performed by me at Caltech, unless explicitly noted in the text or figures. I also
contributed the analysis on the simplified model system illustrating the challenge of retroactivity in
Section 3.2. Detailed first principles models and parameter fitting were performed by J. Kim and
context of transcriptional circuits was also presented in [39].
•Chapter 4: Robust properties of natural networks. As already noted, the molecular
circuitry of living organisms performs remarkably robust regulatory tasks, despite the intrinsic
vari-ability of its components. A large body of research has in fact highlighted that robustness is often
a structural property of biological systems. However, there are few systematic methods to
mathe-matically model and describe structural robustness. With a few exceptions, numerical studies have
been thede facto standard for this type of investigation.
In this chapter I will highlight how robust stability of equilibria in biological networks can be
analyzed using Lyapunov and invariant sets theory. In particular, the analysis is focused on the
structure of ODE models rather than on their specific functional expressions. Without resorting to
extensive numerical simulations, the stability properties of well-known biological networks will be
rigorously proved to be robust. Several case studies will be considered, including thelacoperon and
the mitogen-activated protein kinase (MAPK) pathway.
This project was developed with Prof. Franco Blanchini at the University of Udine. F. Blanchini
and I conceived together the general idea of structural analysis of biological models using Lyapunov
functions. F. Blanchini mainly focused on the technical results; I contributed the models and assessed
the key assumptions and interpretations of the results in a biological context. This chapter reports
Chapter 2
Flux control for biochemical
networks
2.1
Introduction
Cellular pathways rely heavily on a regulated flux of nucleic acids, transcription factors, and other
metabolites. In the era of synthetic biology, it is important to understand and optimize the
mecha-nisms that control and optimize molecular flows. This will contribute to the formulation of systematic
design rules for constructing large biochemical networks [92]. (In the following I will use the words
flux and flow interchangeably.)
Here, I will consider a simple model problem: given two reagents that bind to form a product,
how can we equate their flow through the design of suitable feedback loops? If the two flows are
not matched, we could fall in a scenario where (1) the reagent with the higher flux will accumulate,
creating a potentially harmful excess of such species and (2) the flow of product will be limited by
the lower reagent flux. Two different network design solutions to these problems will be proposed,
both based on the use of feedback. A desirable feature of such designs would be their robustness
(low sensitivity) with respect to the open loop production rate of the reagents.
The first scheme relies on the use of negative auto-regulation: either species in excess is designed
to down-regulate its own production rate. Situation (1) is therefore avoided. The second scheme is
based on positive cross-regulation: if one of the reagents is in excess, it will increase the production
rate of the second reagent. This second architecture aims at avoiding point (2). The main feature
of both these schemes is that feedback is implemented using stoichiometric reactions and without
making time-scale separation arguments, which typically yield Michaelis-Menten or Hill functions.
The flux-matching problem and the outlined solutions will first be described with a simple
sys-tem of ODEs. Then, I will outline how the properties of these feedback schemes can be assessed
experimentally using transcriptional circuits. Experiments on the implementation of the negative
flow-matching is achieved robustly with respect to the open loop rates. On the contrary, the positive
cross-regulatory scheme presents several design challenges and the data currently available do not
verify the flux matching property conclusively.
2.2
Problem formulation
Consider a simple chemical reaction network
T1
β1
*R1+ T1,
T2
β2
*R2+ T2,
R1+ R2 k
*P. (2.1)
Two chemical species T1 and T2 produce, respectively, reactants R1 and R2, at rates β1, β2. The
reactants then bind to form an output product P. T1 and T2 could be, for instance, two genes
whose mRNA or protein outputs R1 and R2 must interact stoichiometrically to form a complex
useful for a downstream process. A pictorial representation of the network is given in Figure 2.1 A.
The differential equation corresponding to the dynamics of Ri is:
d[Ri]
dt =βi·[Ti]−k [Ri][Rj], i,j∈ {1,2},i6= j. (2.2)
The build-up of the product P is clearly conditioned by the ratesβ1, β2 and the concentrations
[T1] and [T2]. If the production rates for R1 and R2 are significantly different, one can make two
observations. First, the reactant produced at the higher rate will accumulate in the system. Second,
the lower production rate becomes a bottleneck for the formation of P. For instance, if [T1][T2], the concentration of R2builds up; at the same time the yield of P is limited by the production rate
of R1. If reactions (2.1) represent a genetic circuit in a cellular host, an excess of R2could harm the
organism, besides causing a waste of resources. Ideally, biochemical or metabolic networks should
include feedback loops able to eliminate excess production of molecules that are not utilized by the
system, and increase insufficient production of molecules in high demand. The solution trajectories
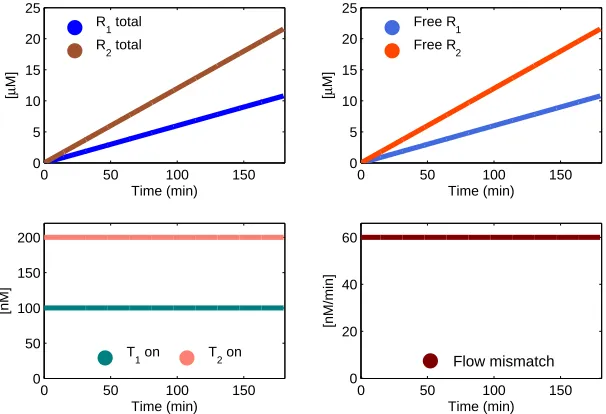
for equation (2.2) are shown in Figure 2.2. Parameters were chosen asβi= 0.01/M, k = 2·103/M/s, T1= 100 nM, T2= 200 nM.
In this work, I will consider the model system (2.1) when the production rates for Ri are not
balanced. The question that will be asked is: If we could design R1 and R2 to interact with the
generating species T1 and T2, could we achieve self-regulation and matching of the flux rates for
the two reactants, robustly with respect to the open loop rates? I will investigate this question by
looking at the effects of the feedback loops that can be generated by R1and R2. In particular, I will
B
A
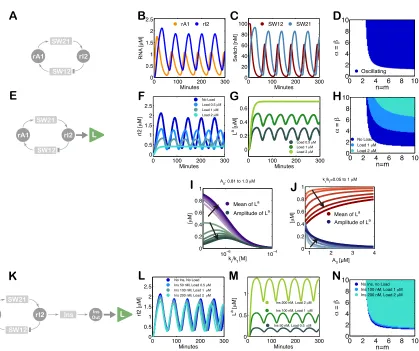
C
T1 R1
R2 T2
P
T1 R1R
2 T2
P [R1]>[R2]
[R2]>[R1]
T1 R1R
2 T2
P [R1]>[R2]
[image:22.612.174.477.248.455.2][R2]>[R1]
Figure 2.1: A. Schematic representation for our model problem (2.1). B. Negative feedback scheme to control the flow of R1 and R2, corresponding to equations (2.3). The comparison between the concentrations of R1 and R2 is implicit, due to the formation of the product P. C. Positive feedback interconnection to control the flow of R1 and R2, corresponding to equations (2.7).
0 50 100 150
0 50 100 150 200
Time (min)
[nM]
T1 on T2 on
0 50 100 150
0 5 10 15 20 25
Time (min)
[
µ
M]
Free R1
Free R 2
0 50 100 150
0 5 10 15 20 25
Time (min)
[
µ
M]
R1 total
R
2 total
0 50 100 150
0 20 40 60
Time (min)
[nM/min]
Flow mismatch
Figure 2.2: Numerical solution to the differential equations (2.2). Bottom right: absolute value of the flux mismatch between the total amount of species Rtot1 and Rtot2 .
(Figure 2.1 C). I will assume that the feedback occurs by mass action chemical reactions.
2.2.1
Self-repression
Free molecules of Ri, i = 1,2, bind to active Ti thereby inactivating it:
Ri+ Ti
δi *T∗i,
T∗i
αi *Ti,
where T∗i is an inactive complex. We assume that Ttot
i = Ti+ T∗i, and that T∗i naturally reverts to
its active state with a first-order rate αi. The total amount of Ri is [Rtoti ] = [Ri] + [T∗i] + [P]. A
differential equations are:
d[Ti]
dt =αi([T tot
i ]−[Ti])−δi[Ri][Ti], d[Ri]
dt =βi[Ti]−k [Ri][Rj]−δi[Ri][Ti]. (2.3)
For illustrative purposes, the above differential equations are solved numerically. The parameters
chosen are: α1=α2= 3·10−4 /s,β1=β2= 0.01 /s,δ1=δ2= 5·102 /M/s, and k = 2·103/M/s. An imbalance in the production rates of R1 and R2 is created by setting [T1](0) = [Ttot1 ] = 100 nM
and [T2](0) = [Ttot2 ] = 200 nM, while [R1](0) = [R2](0) = 0. The overall result of this feedback
interconnection is that the mismatch in the flow rate of R1and R2is reduced, as shown in Figure 2.3.
The flow rate is defined as the derivative of the total amount of [Rtot
i ]. The flow rate mismatch is
defined as the absolute value of the difference between the two fluxes. The effect of changing the
feedback strength, for simplicity chosen as δ1 = δ2, is shown in Figure 2.4: the figure shows the
mean active fraction of [Ti] and the mean flow mismatch over a trajectory simulated for 10 hours.
The mean is shown, rather than steady-state values, to capture the behavior of the system over the
whole trajectory.
It is possible to examine the nullclines relating T1 and ¯T2, and find the equilibria ¯T1and ¯T2 as
intersection of these nullclines:
˙
Ti = 0 =⇒ Ri =
αi(Ttoti −Ti)
δiTi
,
˙
Ri= 0 =⇒ Ri =
βiTi kRj+δiTi
.
To simplify the derivation, we set δ1 = δ2 = δ, β1 = β2 = β, α1 = α2 = α. Equating the two
expressions forRi, we get the following equations (for i= 1,2 andj= 1,2):
α
δ 2
k Ttot
i −Ti Ti
Ttot
j −Tj Tj
!
+α(Ttoti −Ti)−βTi = 0.
We can find an expression of the nullclines by introducing a change of variables u =Ttot1 −T1
T1
and
v =Ttot2 −T2
T2
, and defining φ1 =ψ1 = αδ 2
k, φ2 =αTtot1 , ψ2 =αTtot2 , φ3 =βTtot2 , and finally
ψ3=βTtot1 :
u2(φ1v) + u(φ1v +φ2−φ3 1
1 + v)−φ3 1
1 + v = 0, (2.4)
v2(ψ1u) + v(φ1u +ψ2−ψ3 1
1 + u)−ψ3 1
The roots of the equations above represent the nullclines of the system. Because all the
param-eters in these equations are positive, there is always a single root. The nullclines are numerically
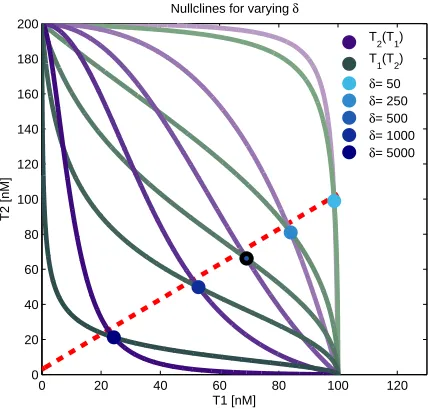
solved, for varyingδ, in Figure 2.5.
A condition for flow matching at steady-state can be derived as follows:
˙
R1−R˙2= 0,
β1T1−δ1T1R1=β2T2−δ2T2R2.
Substituting the expressions for R1 and R2 that can be derived by setting ˙T1= 0 = ˙T2, we get:
β1T¯1−α1(Ttot1 −T¯1) =β2T¯2−α2(Ttot2 −T¯2).
Takingα1=α2=α, β1=β2=β we get:
¯
T2= ¯T1+
α
α+β(T
tot
2 −T
tot
1 ). (2.6)
The flow matching condition above is shown in Figure 2.5, red dashed line. If β α, i.e., the production of Ri is much faster than the generating species Ti inactivation rate, then the condition
can be rewritten as:
¯ T1≈T¯2.
0 50 100 150
0 50 100 150 200
Time (min)
[nM]
T
1 on T2 on
0 50 100 150
0 0.5 1 1.5
Time (min)
[
µ
M]
Free R1
Free R 2
0 50 100 150
0 2 4 6 8 10
Time (min)
[
µ
M]
R1 total
R2 total
0 50 100 150
0 20 40 60
Time (min)
[nM/min]
[image:24.612.174.478.433.656.2]Flow mismatch
100 102 0
50 100 150 200
Mean concentration
δ [/M/s]
[nM]
T1 on
T2 on
100 102
10 20 30 40 50
δ [/M/s]
[nM]/min
[image:25.612.218.434.399.604.2]Mean flow mismatch
Figure 2.4: Numerical simulation for the negative feedback scheme (2.3), showing the mean concentration of active generating species T1and T2and the mean flow mismatch as a function of the feedback parameter rate δ. The points corresponding to the set of nominal parameters (trajectories in Figure 2.6) are circled in black. The mean is taken over a trajectory of 10 hours. The stronger the negative feedback, the less R1 and R2 are produced by the two subsystems.
0 20 40 60 80 100 120
0 20 40 60 80 100 120 140 160 180 200
T1 [nM]
T2 [nM]
Nullclines for varying δ
T
2(T1)
T
1(T2)
δ= 50
δ= 250
δ= 500
δ= 1000
δ= 5000
2.2.2
Cross-activation
Free molecules of Ri bind to inactive Tjand activate it:
Ri+ T∗j
δij
*Tj
Ti
αi *T∗i,
where again T∗i is an inactive complex and Ttot
i = Ti + T∗i. The total amount of Ri is [Rtoti ] =
[Ri]+[Tj]+[P]. We now assume that Tinaturally reverts to its inactive state with rateαi. Figure 2.1
B shows the scheme associated with this feedback interconnection. The corresponding differential
equations are
d[Ti]
dt =−αi[Ti] +δji[Rj]([T tot
i ]−[Ti]), d[Ri]
dt =βi[Ti]−k [Ri][Rj]−δij[Ri]([T tot
j ]−[Tj]). (2.7)
The above differential equations were solved numerically. The parameters were chosen for
il-lustrative purposes as α1 = α2 = 3·10−4 /s, β1 = β2 = 0.01 /s, δ1 = δ2 = 5·102 /M/s, and k = 2·103/M/s. The total amount of templates was chosen as [Ttot
1 ] = 100 nM, [Ttot2 ] = 200
nM. The initial conditions of active [Ti] are set as [T1](0) = 10 nM and [T2](0) = 160 nM, while
[R1](0) = [R2](0) = 0. The overall result of this positive feedback interconnection is shown in
Fig-ure 2.6. The flow rate is defined again as the derivative of the total amount of [Rtoti ]. The flux
mismatch is defined as the absolute value of the difference between the two flow rates. The effect of
changing the feedback strength, where for simplicityδ1=δ2, is shown in Figure 2.7, which plots the
mean active fraction of [Ti] and the mean flow mismatch over a trajectory simulated for 10 hours.
The mean is shown, rather than steady-state values, to capture the behavior of the system over the
whole trajectory. The right panel in Figure 2.7 seems to indicate that the flux mismatch of the two
circuits is minimized for a certain range of δ around the nominal value of δ = 5·102. However, for values of δthat are much smaller or much larger than the nominal value of 5·102, the system dynamics do not reach steady-state within the simulated 10 hours. We will further explore the
behavior of the system’s equilibria and flow matching conditions, as done for the negative feedback
scheme.
The nullclines of the system in the T1-T2 space can be calculated as done for the negative
˙
Tj= 0 =⇒ Ri=
αjTj
δij(Ttotj −Tj)
,
˙
Ri= 0 =⇒ Ri=
βiTi
kRj+δij(Ttotj −Tj)
.
To simplify the derivation, we set δ12 =δ21=δ,β1=β2=β, α1 =α2 =α. Equating the two
expressions for Ri, we get the following equations (for i = 1,2 and j = 1,2):
α
δ 2
k
T
i Ttot
i −Ti
T
j Ttot
j −Tj !
+αTi−βTj= 0. (2.8)
We can find an expression of the nullclines by introducing a change of variables z = T1
Ttot 1 −T1
and
w = T2
Ttot 2 −T2
, and defining φ1 =ψ1 = αδ 2
k,φ2 =αTtot1 ,ψ2 =αTtot2 , φ3 =βTtot2 , and finally
ψ3=βTtot1 :
z2(φ1v) + z(φ1w +φ2−φ3 w
1 + w)−φ3 w
1 + w= 0, (2.9)
w2(ψ1z) + w(φ1z +ψ2−ψ3 z
1 + z)−ψ3 z
1 + z = 0. (2.10)
The roots of the equations above represent the nullclines of the system. Because all the
param-eters in these equations are positive, there is always a single root. The nullclines are numerically
solved, for varyingδ, in Figure 2.8.
A condition for flow matching at steady-state can be derived as follows:
˙
R1−R˙2= 0,
β1T1−δ21R1(Ttot2 −T2) =β2T2−δ12R2(Ttot1 −T1).
Substituting the expressions for R1 and R2 that can be derived by setting ˙T1= 0 = ˙T2, we get:
β1T¯1−
δ21
δ12
α2T¯2=β2T¯2−
δ12
δ21
α1T¯1.
Takingα1=α2=α, β1=β2=β, and δ12=δ21=δwe get:
¯
T2= ¯T1. (2.11)
rate for the generating species) or increasingδ(speed of the positive feedback), with respect to the
nominal values chosen here, causes the equilibrium of the system to be pushed toward the upper right
corner of Figure 2.8. Moreover, when decreasing αor increasing δthe system reaches equilibrium
on a timescale in the order of several dozens of hours. Explicit tradeoffs on the effects of αand δ
may be found by further analysis on the nullclines and on the locus of equilibria in equation (2.8).
0 100 200 300
0 50 100 150 200
Time (min)
[nM]
T1 on T2 on
0 100 200 300
0 0.5 1 1.5 2 2.5
Time (min)
[
µ
M]
Free R 1 Free R
2
0 100 200 300
0 5 10 15
Time (min)
[
µ
M]
R
1 total
R
2 total
0 100 200 300
0 20 40 60 80
Time (min)
[nM/min]
Flow mismatch
100 102 104 0
50 100 150 200
Mean concentration
δ [/M/s]
[nM]
T1
T2
100 102 104
0 5 10 15 20
δ [/M/s]
[nM]/min
[image:29.612.218.435.420.622.2]Mean flow mismatch
Figure 2.7: Numerical simulation for the positive feedback scheme model (2.7), showing the mean concentration of active generating species T1 and T2 and the mean flux mismatch as a function of the cross-activation rate parameter rateδ. The points corresponding to the set of nominal parameters (trajectories in Figure 2.6) are circled in black. The mean is taken over a trajectory of 10 hours. The flux mismatch of the two circuits seems to be minimized for a certain range of δ around the nominal value of δ = 5·102. However, for values of δ that are much smaller or much larger than the nominal value of 5·102, the system dynamics do not reach steady-state within the simulated 10 hours. Figure 2.8 shows the numerically computed nullclines of the system and the corresponding equilibria for varyingδ.
0 20 40 60 80 100 120
0 20 40 60 80 100 120 140 160 180 200
T1 [nM]
T2 [nM]
Nullclines for varying δ
T
2(T1)
T
1(T2)
δ= 50
δ= 250
δ= 500
δ= 1000
δ= 5000
2.3
Implementation with transcriptional circuits
Repression or activation
! Repression or
activation
!
! !
! !
Genelet 1
Genelet 2
RNA 2 RNA 1
RNAP
RNAP !
RNAP
!
RNAP
Figure 2.9: Scheme highlighting the general idea behind the transcriptional circuits implemen-tation of the two feedback interconnections shown in Figures 2.1 B and C. Two RNA species bind to form a product, and their regulatory domains are sequestered. The feedback loops are active when either species is in excess, and therefore its regulatory domains are not covered.
The model problem described above can be experimentally tested using transcriptional circuits.
The two species T1 and T2 correspond to two switches, whose RNA transcripts are the output
reagents R1 and R2. Such transcripts are designed to bind and form an RNA complex P. Since the
focus of this work is the investigation of the effects of feedback, the structure of P and its functionality
as a standalone complex will be neglected. Depending on the feedback scheme to be implemented,
the RNA species R1 and R2 will be designed to have different domains. However, once R1 and R2
are bound and form P, it will be required that the complex is inert and all the regulatory domains
for negative auto-regulation or cross-activation are covered. This idea is depicted in Figure 2.9.
2.3.1
Self-repression
A graphical sketch of the domain-level design for the self-repression interconnection is shown in
Fig-ure 2.10 A. The RNA outputs of each genelet are designed to be complementary to the corresponding
activator strand. However, the two RNA species are also complementary. This specification on the
design of the transcripts introduces a binding domain between Tiand Rj, which is considered another
off state, as shown in Figure 2.10 B. Such complex is a substrate for RNase H and the RNA strand is
degraded by the enzyme, releasing the genelet activation domain. We assume that the transcription
efficiency of an RNA-DNA promoter complex is very low. This hypothesis was not experimentally
challenged for this specific system; however, in Section 3.7.14 we show that this assumption is valid
for other genelets with the same promoter domain.
here, with two-domain RNA transcripts, was originally presented in [40]. ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! ! R1 R2 P T1 on
T1 off T2 off
T2 on A2 A1 R2•A2 R1•A1 RNAP RNaseH RNaseH RNAP
A
B
! ! ! ! ! ! R1 T2 off RNaseH ! ! ! ! R2 ! ! T1 off RNaseH a1’ a1 t1 a1’ t1’ a2 t2a2 t2 a1 t1 a2’ t2’ a2’ a1’ t1’ a1
t1 t2 a2
a2’ t2’ a1’ t1’ a2 t2 a1 t1 a2’ t2’ a1 t1 a2 t2 a1’ a2’ a1 t1 a2’
t2’ t1’ a1’ a2 t2
a2 t2 a2’ a1 t1 a1’
[image:31.612.113.542.99.391.2]T1 off T2 off
Figure 2.10: General reaction scheme representing a transcriptional circuit implementation of the negative feedback scheme in Figure 2.1 B. Complementary domains have the same color. Promoters are in dark gray, terminator hairpin sequences in light gray. The RNA output of each genelet is designed to be complementary to its corresponding activator strand. The two RNA species are also complementary. A. Desired self-inhibition loops. B. Undesired cross-hybridization and RNase H mediated degradation of the RNA-template complexes.
2.3.1.1 Modeling
Based on the outlined design specifications and the resulting molecular interactions, we can build a
model for the system. The switches Ti and Tj can have three possible states: the on state where
activator and template are bound and form the complex TiAi; the off state given by free Ti; the off
state represented by Rjbound to Ti forming TiRj. An off state still allows for RNAP weak binding
and transcription. Throughout this derivation, the dissociation constants are omitted when assumed
to be negligible. It is hypothesized that the concentration of enzymes is considerably lower than that
The overall reactions are, for i∈ {1,2},j∈ {2,1}:
Activation Ti+ Ai
kTiAi
* Ti·Ai
Inhibition Ri+ Ti·Ai
kRiTiAi
* Ri·Ai+ Ti
Annihilation Ri+ Ai
kRiAi
* Ri·Ai
Output formation Ri+ Rj
kRiRj
* Ri·Rj
Undesired hybridization Rj+ Ti
kRjTi
* Rj·Ti.
The enzymatic reactions are, for i∈ {1,2},j∈ {2,1}:
Transcription: on state RNAP + Ti·Ai k+ONii
* )
k−ONii
RNAP·Ti·Ai kcatONii
* RNAP + TiAi+ Ri
Transcription: off state RNAP + Ti k+OFFii
* )
k−OFFii
RNAP·Ti kcatOFFii
* RNAP + Ti+ Ri
Transcription: off state, undesired RNAP + Rj·Ti k+OFFji
* )
k−OFFji
RNAP·Rj·Ti
kcatOFFji
* RNAP + Rj·Ti+ Ri
Degradation RNaseH + Ri·Ai
k+Hii
* )
k−Hii
RNaseH·Ri·Ai kcatHii
* RNaseH + Ai
RNaseH + Rj·Ti k+Hji
* )
k−Hji
RNaseH·Rj·Ti kcatHji
* RNaseH + Ti.
Given the above reactions, it is straightforward to derive a set of ODEs as follows:
d
dt[Ti] =−kTiAi[Ti] [Ai] + kRiTiAi[Ri] [Ti·Ai]−kRjTi[Rj] [Ti] + kcatHji[RNaseH·Rj·Ti], d
dt[Ai] =−kTiAi[Ti] [Ai]−kRiAi[Ri] [Ai] + kcatHii[RNaseH·Ri·Ai], d
dt[Ri] =−kRiRj[Ri] [Rj]−kRiTiAi[Ri] [Ti·Ai]−kRiTj[Ri] [Tj]−kRiAi[Ri] [Ai]
+ kcatONii[RNAP·Ti·Ai] + kcatOFFii[RNAP·Ti] + kcatOFFji[RNAP·Rj·Ti], d
dt[Ri·Rj] = + kRiRj[Ri] [Rj], d
dt[Rj·Ti] = + kRjTi[Rj] [Ti]−kcatHji[RNaseH·Rj·Ti].
(2.12)
The molecular complexes that appear in the right-hand side of the above equations can be expressed
We assume that binding of enzymes to their substrate is faster than the subsequent catalytic step,
and that the substrate concentration is much larger than the amount of enzyme. This allows us to
use the standard Menten quasi-steady-state expressions. We need to define the
Michaelis-Menten coefficients: for instance, for the ON state of the template, define: kMONii=
k−ONii+kcatONii
k+ ONii
.
Then the following expressions hold:
[RNAPtot] =[RNAP]
1 +[T1·A1]
kMON11
+ [T1]
kMOFF11
+[T2·A2]
kMON22
+ [T2]
KMOFF22
+[R2·T1]
kMOFF21
+[R1·T2]
kMOFF12
,
[RNaseHtot] =[RNaseH]
1 +[R1·A1] kMH11
+[R2·A2] kMH22
+[R2·T1] kMH21
+[R1·T2] kMH12
.
We can easily rewrite the above equations as [RNAP] = [RNAPP tot] and [RNaseH] = [RNaseHH tot], with
a straightforward definition of the coefficients P andH. Finally:
[RNAP·Ti·Ai] =
[RNAPtot] [Ti·Ai] P·kMONii
,
[RNAP·Rj·Ti] =
[RNAPtot] [Rj·Ti] P·kMOFFji
,
[RNAP·Ti] =
[RNAPtot] [Ti] P·kMOFFii
,
[RNaseH·Ri·Ai] =
[RNaseHtot] [Ri·Ai] H·kMHii
,
[RNaseH·Rj·Ti] =
[RNaseHtot] [Rj·Ti] H·kMHji
,
which can be substituted in equations (2.12).
The nonlinear set of equations (2.12) is analyzed numerically. The parameter values used in these
simulations are reported in Table 2.1. Such parameters are consistent with those in [63]; this is a fair
assumption since the design of this system is essentially identical to that of a repressible switch. For
simplicity we assume that the circuits are symmetric, and their parameters are therefore identical.
We can assess the performance of the circuit by just creating an imbalance in the concentration of the
templates. Figure 2.11 shows the system trajectories that correspond to zero initial conditions for
[Ai] and [Ri], while the complexes [T1A1] = [Ttot1 ] = 100 nM, [T2A2] = [Ttot2 ] = 50 nM, [Atot1 ] = 100
nM and [Atot
2 ] = 50 nM. (The simulation first allows for equilibration of all the DNA strands in the
absence of enzymes. Only the portion of trajectories after addition of enzymes is shown.) The total
concentration of enzymes is assumed to be [RNAPtot] = 80 nM and [RNaseHtot] = 8.8 nM. The
RNAP and RNase H concentrations were chosen based on typical experimental conditions. (For a
brief discussion on estimating enzyme concentrations, see Table 3.3, Section 3.7.4.) Note that the
concentration of RNAP is not negligible relative to the total amount of genelets present: this means
that the Michaelis-Menten approximation may not be accurate in this case. The simulation results
shown at Figure 2.3.
Table 2.1: Simulation Parameters for Equations (2.12)
Units: [1/M/s] Units: [1/s] Units: [M]
kTiAi = 4·10
4
kcatONii= 0.06 kMONii = 250·10
−9
kTiAiRi= 5·104 kcatOFFii = 1·10−3 kMOFFi = 1·10−6
kAiRi = 5·10
4
kcatOFFij = 1·10
−3
kMOFFij = 1·10 −6
kRiTj = 1·10
3
kcatHii=.1 kMHii= 50·10 −9
kRiRj = 1·10
6
kcatHji=.1 kMHji = 50·10 −9
0 100 200 300
0 20 40 60 80 100 Time (min) [nM] T
1 on T2 on
0 100 200 300
0 0.02 0.04 0.06 0.08 Time (min) [ µ M] Free R 1 Free R 2
0 100 200 300
0 2 4 6 8 Time (min) [ µ M] R
1 total
R
2 total
0 100 200 300
[image:34.612.152.499.201.482.2]−2 0 2 4 6 8 10 Time (min) [nM/min] Flow mismatch
Figure 2.11: Numerical simulation for equations (2.12). Parameters are chosen as in Table 2.1. [T1A1] = [Ttot1 ] = 100 nM, [T2A2] = [Ttot2 ] = 50 nM, [Atot1 ] = 100 nM, and [Atot2 ] = 50 nM, [RNAPtot] = 80 nM, and [RNaseHtot] = 8.8 nM. These results are consistent with those of the simple model proposed in equations (2.3), and analyzed numerically in Figure 2.3.
2.3.1.2 Experimental results
We expect the feedback scheme to downregulate the production of either RNA species when in excess
with respect to the other. For instance, if the concentration of [T1·A1] is twice the concentration of [T2·A2], the concentration of R1produced will clearly exceed that of R2. If the feedback scheme is working correctly, we expect to notice a decrease in the percentage of template [T1·A1]. We can easily verify this hypothesis by labeling the 5’ end of the non-template strand of the genelets
with different fluorescent dyes, and by labeling the corresponding activator strand with a quencher
templates will be quenched. For instance, when A1is stripped off T1, the T1fluorescence signal will
increase. For convenience, the fluorescence traces will be processed to map the measured signal to
the corresponding active genelet concentrations. In all the fluorescence traces shown here, the total
amount of activators is stoichiometric to the total amount of templates: [Atot
i ] = [Ttoti ].
Figure 2.12 A shows the behavior of the two genelets in isolation: we can verify that each
genelet self-inhibits after the enzymes are added. (For details on the data normalization procedure,
refer to Section 2.3.1.3.) The concentration of RNA present in solution can be measured through gel
electrophoresis, as shown in Figure 2.12 B: lanes 1 and 2 show that transcription is effectively absent.
When the two genelets are present in solution in stoichiometric amount, their RNA outputs bind
quickly to form a double-stranded complex, and therefore the feedback loops become a secondary
reaction (by design thermodynamically less favorable than the R1·R2complex formation). As shown in Figure 2.12 C, the two genelets only moderately self-repress. The total RNA concentration in
solution is high, as shown in the denaturing gel in Figure 2.12 B, lanes 3 and 4. A discussion on the
accuracy of the gel data is in Section 2.3.1.3.
When the templates [Ttot
1 ] and [Ttot2 ] are in different ratios, the system behavior is shown in
Figure 2.13 A. We can plot the resulting initial active template ratio (which corresponds to the total
template ratio) versus the steady-state one: we find that the system behaves symmetrically and the
steady-state ratio is close to one across all the initial ratios. Therefore, given open loop transcription
rates that differ across a factor of 1–3, these results suggest that the system robustly matches the
flux of R1 and R2. If the concentration of [Ttoti ] and [A tot
i ] is changed over time, the steady-state
concentration of active genelets adjusts as shown in Figure 2.14 A and B. Samples from this set of
experiments were analyzed using a denaturing gel: the results are shown in Figure 2.14 C and D
(corresponding to the traces in Figure 2.14 A and B, respectively) and show the total RNA amount
in solution and that [Rtot
1 ]≈[Rtot2 ], as desired (Figure 2.14 E and F).
The data in Figure 2.13 A were fitted using MATLAB, restricting the search algorithm to optimize
a subset of parameters that are shown in Table 2.2. This subset of parameters was chosen to assess
whether varying the branch migration rates and the enzyme speeds could satisfactorily explain the
data collected. Such parameters were used to numerically compute equations (2.12), generating the
simulated time traces shown in dashed lines in Figures 2.12 and 2.13. The fitted parameters differ
from the initially postulated parameters: in particular, the binding rates for activation, inhibition,
and output formation are much faster than what initially was assumed (Table 2.1); in particular,
the fitted output formation rate is too high and not physically acceptable. Clearly, the current fits
may be improved by extending the parameter space; this will be part of the future work on this
0 50 100 150 −50
0 50 100 150
Concentration (nM)
Time (min) [T1](0)=100 nM [T2](0)=0 nM
0 50 100 150
−50 0 50 100 150
Concentration (nM)
Time (min) [T1](0)=0 nM [T2](0)=100 nM
0 50 100 150 200
0 20 40 60 80 100
Concentration (nM)
Time (min) [T1](0)=100 nM [T2](0)=100 nM T1 100nM
T2 100nM T1
100nM T2 100nM
100 150 200 300
90
80
70
60
A
B
C
R1
R2
1 2 3 4
[image:36.612.110.542.95.311.2]Act RNAP
Figure 2.12: A. Experimental data showing the isolated active genelet concentrations as a function of time: the self-inhibition reaction turns the switches off, and the RNA concentration in solution is negligible, as verified in the gel electrophoresis data in panel B, lanes 1 and 2 (samples taken at steady-state after 2 h). Dashed lines represent numerical trajectories of equations (2.12), using the fitted parameters in Table 2.2. B. Denaturing gel image: lanes 1 and 2 show that the switches in isolation self-inhibit and no significant transcription is measured. Lanes 3 and 4 show the total RNA amount in samples from the experiment shown at panel C, taken at steady-state after 2 h. When the genelets are in stoichiometric amount, their flow rates are already balanced and there is only a moderate self-inhibition.
Table 2.2: Fitted Parameters for (2.12)
.
Units: [1/M/s] Units: [1/s]
kTiAi = 2.9·105 kcatONii= 0.06
kTiAiRi= 5·10
5
kcatHii=.09
kAiRi = 5·10
4
kcatHji=.09
kRiTj = 1·10
3
kRiRj = 2·10
[image:36.612.249.398.581.676.2]0 50 100 150 200 0 20 40 60 80 100 Concentration (nM)
Time (min) [T1](0)=50 nM [T2](0)=100 nM
0 50 100 150 200 0 20 40 60 80 100 Concentration (nM)
Time (min) [T1](0)=100 nM [T2](0)=50 nM
0 50 100 150 200
0 50 100 150 200 Concentration (nM)
Time (min) [T1](0)=100 nM [T2](0)=200 nM
0 50 100 150 200
0 50 100 150 200 Concentration (nM)
Time (min) [T1](0)=200 nM [T2](0)=100 nM
0 50 100 150 200 0
100 200 300
Concentration (nM)
Time (min) [T1](0)=100 nM [T2](0)=300 nM
0 50 100 150 200
0 100 200 300
Concentration (nM)
Time (min) [T1](0)=300 nM [T2](0)=100 nM
A
B
0 1 2 3 4
0 1 2 3 4
Final ratio of active genelets
Initial ratio of active genelets
Numerical Model No feedback
Data: T1 Varied, T2 Fixed Data: T2 Varied, T1 Fixed
0 100 200 300
0 20 40 60 80 100 Concentration (nM)
Time (min) [T1](0)=100 nM [T2](0)=100 nM Act RNAP



![Figure 2.11: Numerical simulation for equations (2.12). Parameters are chosen as in Table 2.1.[T1A1] = [Ttot1 ] = 100 nM, [T2A2] = [Ttot2 ] = 50 nM, [Atot1 ] = 100 nM, and [Atot2 ] = 50 nM,[RNAPtot] = 80 nM, and [RNaseHtot] = 8.8 nM](https://thumb-us.123doks.com/thumbv2/123dok_us/8814208.919732/34.612.152.499.201.482/figure-numerical-simulation-equations-parameters-chosen-rnaptot-rnasehtot.webp)