1071-412X/96/$04.0010
Copyrightq1996, American Society for Microbiology
Production of T-Helper Cell Subsets and Cytokines by
Lymphocytes from Patients with Chronic
Mucocutaneous Candidiasis
LISA J. KOBRYNSKI,1* LISA TANIMUNE,1,2LAURIE KILPATRICK,1,2DONALD E. CAMPBELL,1,2,3
ANDSTEVEN D. DOUGLAS1,2,3
Division of Allergy, Immunology, and Infectious Diseases,1Joseph Stokes Jr. Research Institute,2and Clinical
Immunology Laboratory,3Department of Pediatrics, The Children’s Hospital of Philadelphia, University of
Pennsylvania School of Medicine, Philadelphia, Pennsylvania 19104
Received 29 December 1995/Returned for modification 25 March 1996/Accepted 29 July 1996
Chronic mucocutaneous candidiasis (CMC) is a heterogeneous group of disorders characterized by recur-rent and persistent superficial candidal infections. Cytokine-induced dysregulation of T-helper cell function has been described in other immune-deficient states but has not been studied in CMC patients. We studied T-helper cell subsets by flow cytometry and cytokine production by stimulated lymphocytes in six CMC patients, two healthy pediatric controls, and five healthy adult controls. Peripheral blood lymphocytes were
stimulated in vitro with phytohemagglutinin orCandida albicansextract, and the production of interleukin-2R
(IL-2R), IL-4, IL-10, and gamma interferon in the supernatants was measured by enzyme-linked
immunosor-bent assay. CMC patients had a decrease in the CD41/CD291cell population compared with the numbers in
controls (P< 0.02). The percentage of CD41/CD45RA1cells was greater in patients than in controls, but the
difference was not significant. There was no difference in the production of IL-10 or gamma interferon by the
patient lymphocytes. CMC patients produced more IL-4 than the controls (P< 0.001), whereas the controls
tended to produce more IL-2R than the patients (P50.19). These findings support the concept that a decrease
in CD41/CD291T-helper inducer cells along with T-helper cell dysregulation may lead to defective memory
responses to antigens in CMC patients and a decrease in cell-mediated immunity due to inhibition of TH1 cells
by increased levels of IL-4.
Chronic mucocutaneous candidiasis (CMC) is a heteroge-neous group of disorders characterized by recurrent and per-sistent candidal infections of the mucosa, nails, and skin, usu-ally with Candida albicans (16). Patients with CMC rarely develop disseminated or invasive infections with candida or other microorganisms (2, 6), but some patients have been observed to be susceptible to infection with organisms such as
Staphylococcus aureus,Mycobacterium avium-M. intracellulare,
Histoplasma capsulatum,Cryptococcus neoformans, herpes sim-plex virus and herpes zoster virus (13–15).
CMC is also associated with endocrinopathies and autoim-mune phenomena, such as hypoparathyroidism, hypothyroid-ism, and autoimmune hemolytic anemia. Thus, the clinical manifestations are varied and many different clinical variants have been described (2, 11). We have recently reviewed the occurrence of autoimmune hemolytic anemia in CMC patients (21), and data for four of these patients are included in this report. CMC has also been classified according to the immu-nologic profiles of the patients (16).
The primary immune defect in CMC is thought to lie in the T-cell response toC. albicansantigens (6, 16). Despite the wide variety of clinical manifestations seen in patients with CMC, these patients tend to share certain specific defects in immune function. Lymphocytes from patients with CMC usually have diminished proliferative responses to C. albicans as well as cutaneous anergy to C. albicans. Most patients have normal proliferative responses to phytohemagglutinin (PHA) and
pro-duce normal serum immunoglobulins and normal serum anti-body titers to other antigens, such as diphtheria and tetanus antigens (2). Durandy et al. (10) demonstrated that infection withC. albicansleads to an accumulation of mannan (a Can-dida polysaccharide antigen). This causes activation of sup-pressor T cells capable of inhibiting both the proliferation and the activation of T-helper cells (10). In some patients a plasma inhibitor interferes with lymphocyte responses toC. albicans
antigens (33). Abnormal or inappropriate lymphokine produc-tion by C. albicans-stimulated T cells may play a role in the pathogenesis of CMC (16).
Recently, much attention has been focused on T-cell pro-duction of lymphokines and on how these lymphokines direct the differentiation of TH0 cells into TH1 or TH2 cells, which
promote cell-mediated immune responses and humoral im-mune responses, respectively. Cytokine-induced dysregulation of T-helper cell function has been described in other disease states such as human immunodeficiency virus infection (7, 8) and leishmaniasis (23). In the invasive form of leishmaniasis, inappropriate production of interleukin-4 (IL-4) appears to cause a switch in THlymphocytes from a predominant type 1
response to a predominant type 2 response, leading to aug-mented humoral responses, cutaneous anergy, and impairment of cellular immunity. Human immunodeficiency virus-positive, asymptomatic patients with impaired responses in vitro to re-call antigens have a decreased level of IL-2 production with an elevation in the level of IL-4 production. This switch to a TH2
pattern of cytokine production precedes both the drop in CD4 counts and the onset of symptoms and may herald disease progression (7).
CD4 cells may be segregated into subsets not only by their expression of cytokines but also by their expression of surface * Corresponding author. Present address: Division of
Allergy/Immu-nology/Rheumatology, Department of Pediatrics, Emory University School of Medicine, 2040 Ridgewood Dr. NE, Atlanta, GA 30322. Phone: (404) 727-3380. Fax: (404) 727-3757.
740
on August 17, 2020 by guest
http://cvi.asm.org/
antigens which can be identified by monoclonal antibodies. CD41/CD45RA1cells respond poorly to recall antigens and are thought to represent naive or unprimed CD4 cells. These cells are also called suppressor-inducer cells because of their ability to induce suppressor cell function (19). CD41/ CD45Ro1 cells respond strongly to recall antigens and can induce immunoglobulin production by B cells; thus, they rep-resent a subset of memory T cells (5). CD29 cells are capable of inducing B cells to produce immunoglobulin in response to antigen-specific stimuli or recall antigens (20). CD29 and CD45Ro are coexpressed in healthy donors (19), and the ex-pression of CD29 on CD4 cells correlates with memory or T-helper inducer cell function.
Several investigators have examined the distribution of TH
lymphocytes expressing surface markers characteristic of naive (CD45RA) and memory (CD29) cells in various diseases, such as ataxia-telangiectasia, common variable immune deficiency, X-linked agammaglobulinemia (XLA), mixed connective tis-sue disease, multiple sclerosis, and atopic dermatitis (3, 9, 22, 24, 25, 29). In one study, patients with common variable im-mune deficiency had an increase in the number of CD41/ CD45Ro1cells, leading to speculation that failure of antibody synthesis or a lack of B lymphocytes leads to an expansion in the number of memory CD41/CD45Ro1cells (25). Other in-vestigators have reported a decrease in CD41/CD45RA1cell numbers in these patients (18). In contrast, patients with XLA were found to have a decrease in the number of CD41/ CD45Ro1cells in their peripheral blood, suggesting that the development of memory T cells requires the presence of B cells in order for there to be bidirectional communication between T and B cells (9, 25). Although the regulatory mech-anisms controlling the expression of CD45RA and CD29 on CD4 cells are not known, it is clear that their level of expres-sion is altered in many disease states.
To date, the role of TH1 and TH2 cells and the expression of
CD45RA and CD29 on CD4 cells in CMC has not been stud-ied. An inappropriate response toC. albicansis the hallmark of this disease, which is likely to be a reflection of an imbalance in the TH1 and TH2 responses to this specific antigen. However,
this imbalance must have broader effects in order to explain the other manifestations of this disease. In an effort to gain
some insight into possible aberrant immunoregulatory mecha-nisms operative in this disease, we examined six patients with CMC, with various manifestations of the disease, to determine if they display abnormalities in the composition of their CD41/ CD29 and CD41/CD45RA cell populations which could ex-plain some of the defects seen in this disease. We also exam-ined the cytokine production by stimulated lymphocytes in vitro to determine if it differs from that in healthy controls and if the function of TH1 and/or TH2 cells is defective in patients
with CMC.
MATERIALS AND METHODS
We studied six patients, all of whom were followed in our Immunology Clinic for Chronic mucocutaneous candidiasis. All patients had a history of persistent or recurrent superficial mucocutaneous fungal infections without evidence of specific T-cell disorders such as severe combined immunodeficiency, AIDS, or DiGeorge syndrome. All patients had received in the past intermittent therapy with oral antifungal agents. Three of the six patients have also developed endo-crinopathies and autoimmune disorders such as hypoparathyroidism, hypothy-roidism, and autoimmune hemolytic anemia (Table 1).
Peripheral blood lymphocytes (PBLs) were also obtained from five healthy adult donors, two healthy pediatric patients, and the father of one of the patients, who had persistent onychomycosis but no other features of CMC. This study was approved by the Institutional Review Board of the Children’s Hospital of Phil-adelphia.
Patient and control blood.After consent was obtained, venous blood was collected in heparinized syringes. Peripheral blood mononuclear cells were ob-tained by centrifugation on Ficoll-Hypaque. The cells were washed twice with RPMI 1640 (Gibco, Grand Island, N.Y.), the number of viable lymphocytes was determined by trypan blue staining, and the cells were counted on a hemocy-tometer. The cells were resuspended in RPMI 1640 with 20% fetal calf serum (FCS) at a concentration of 105cells per ml.
Mitogen and antigen proliferation.Mitogen proliferation assays were per-formed by incubating 105PBLs in RPMI 1640 supplemented with penicillin (100 U/ml), streptomycin (100mg/ml),L-glutamine, and 20% FCS. The cells were
cultured in triplicate for 72 h with PHA (1 mg/ml; Sigma, St. Louis, Mo.) at 378C in 5% CO2. After 72 h they received a 6-h pulse with 0.5mCi of [3H]thymidine (Sigma) and were then harvested and washed on glass filters. [3H]thymidine incorporation was measured in a liquid scintillation counter (Beckman).
Antigen proliferation assays were also performed in triplicate with 23105 PBLs per microwell cultured withC. albicansskin test extract (Hollister-Stier, Spokane, Wash.) which had been dialyzed against RPMI 1640, filter sterilized, and titrated against control PBLs. An optimal dilution of 1:4 was used in all subsequent studies. Cells were cultured for 5 to 7 days at 378C in 5% CO2. At the end of the incubation the cells received a 6-h pulse of [3H]thymidine and were then harvested and counted as described above.
Assays for IL-2R, gamma interferon (IFN-g), IL-10, and IL-4.PBLs were TABLE 1. Clinical characteristics of study patients with CMC
Patient no. Age (yr) Sexa Symptoms at presentation Clinical complications Skin testing Autoantibodies
1 22 F Chronic oral and cutaneous candidiasis
Hypothyroidism, autoimmune hemolytic anemia
Candidasp. negative, tetanus positive
ANAb
2 19 F Chronic oral and cutaneous candidiasis
Gastrointestinal malabsorption
NDc
3 8 F Chronic oral and cutaneous
candidiasis
Cyclic neutropenia, sinusitis Candidasp. positive, repeat negative
Smooth muscle
4 22 M Chronic candidal infections of skin and nails
Autoimmune hemolytic anemia, bronchiectasis, hypothyroidism; the patient died of respiratory failure secondary to disseminated candidiasis
Candidasp. negative, tetanus negative
5 10 F Oral and cutaneous
candidal infections
Hypoparathyroidism, alopecia totalis, asthma, rheumatic fever
Candidasp. negative, Trichophytonsp. positive
Mitochondrial parietal cell
6 3 F Chronic candidal infections of skin, nails, and mucous membranes
Hypoparathyroidism ND Mitochondrial
parietal cell
a
F, female; M, male.
b
ANA, antinuclear antibody.
c
ND, not done.
on August 17, 2020 by guest
http://cvi.asm.org/
cultured in triplicate in 24-well flat-bottom culture plates (Falcon) at 53105 cells per well. PBLs were either unstimulated or stimulated with PHA (1 mg/ml) orC. albicansextract. Cells were cultured at 378C in 5% CO2; and the superna-tants were collected at 24, 48, or 72 h, spun at 1,200 rpm to remove debris, and frozen at2708C.
(i) IL-2R assay.IL-2R in the supernatant was measured by an enzyme-linked immunosorbent assay (ELISA) (Predicta; Genzyme, Cambridge, Mass.). The plates were coated with mouse monoclonal antibody to IL-2R, and 75ml of anti-IL-2R biotinylated antibody was added to all the wells. Then, 25ml of each IL-2R standard and sample was added to the appropriate wells. The plate was incubated for 1 h at 378C. Subsequently, the plate was washed five times with a buffered detergent solution, 100ml of IL-2R horseradish peroxidase-labeled streptavidin was added to each well, and the plates were incubated for 15 min at 378C. After washing, 100ml of the tetramethylbenzidine substrate reagent was added for 10 min at room temperature. A total of 100ml of 1 M sulfuric acid was added to stop the reaction, and the absorbance of each well was read on an ELISA plate reader at 450 nm. Values were calculated from a standard curve for recombinant human IL-2R.
(ii) IFN-gassay.The amount of IFN-gpresent in the supernatant at 72 h of culture was determined by using a human IFN-gELISA kit (BioSource Inter-national, Camarillo, Calif.). Briefly, 100 ml of supernatant or human IFN-g standard was added in duplicate to each well of a plate precoated with anti-human IFN-gmonoclonal antibody, and the plate was incubated for 1 h at 248C. The plate was then washed once with a buffered detergent wash solution, 100ml of a rabbit polyclonal anti-human IFN-gantibody was added to each well, and the plate was incubated for 1 h at 248C. The plate was washed, 100ml of an anti-rabbit antibody conjugated to horseradish peroxidase (HRP) was added to each well, and the plate was incubated for 1 h. After washing four times, an HRP substrate solution was added for 1 h. The reaction was stopped by adding 5% sulfuric acid to each well, and the plate was read at 490 nm. The values were calculated by comparison with the standard curve.
(iii) IL-10 assay.IL-10 production was measured by using an IL-10 ELISA kit (BioSource). For this assay, supernatant was collected at 48 h, and the samples were incubated in plates precoated with anti-human IL-10 monoclonal antibody with biotin-labeled anti-human IL-10 for 2 h at 378C. The plate was then washed four times, an HRP-streptavidin conjugate was added, and the plate was incu-bated for 45 min. Then, a substrate solution of tetramethylbenzidine was added for 30 min and the reaction was stopped. The plate was read in an ELISA reader at 490 nm, and values were calculated from a standard curve.
(iv) IL-4 assay.IL-4 was measured in cell culture supernatants at 48 h by using an IL-4 ELISA kit (Genzyme). A 96-well plate was coated with mouse mono-clonal anti-human IL-4 and incubated at 48C overnight. The plate was then washed with a phosphate-buffered saline–Tween solution, the IL-4 standards and supernatants were applied to the plate, and the plate was incubated for 2 h at room temperature. After washing, 100ml of polyvalent rabbit anti-human IL-4 was added to each well and the plate was again incubated for 2 h at room temperature. The plate was washed, biotin-conjugated goat anti-rabbit immuno-globulin was added, and the plate was incubated for 45 min. After washing, 100
ml of streptavidin-conjugated HRP was added to each well for 40 min. The plate was washed one more time. The substrate reagent (chromogen with peroxidase) was then added to each well. The reaction was allowed to proceed for 10 min and was then stopped by the addition of 1 M H2SO4to each well; theA490was read, and the values were calculated from the standard curve.
T-cell phenotyping.Peripheral blood from patients and controls was used for measurement of CD4, CD8, CD41/CD291, and CD41/CD45RA1cell popula-tions. Samples were labeled with fluorescein isothiocyanate-conjugated clonal antibody against CD4 and CD8 and a phycoerythrin-conjugated mono-clonal antibody against CD29 and CD45RA (Coulter Immunology, Hialeah, Fla.). The samples were processed, and the T-cell subsets were quantitated by flow cytometry (EPICS Elite; Coulter Immunology, Hialeah, Fla.).
RESULTS
Mitogen and antigen proliferation.The six patients studied
ranged in age from 3 to 22 years and had various manifesta-tions of CMC, as indicated in Table 1. All patients tested were anergic on delayed-type hypersensitivity skin testing with C. albicansextract. Five of the six patients had a normal lympho-cyte proliferative response to PHA, with a proliferative re-sponse that was at least 69% that of the control subjects. Patient 4 had a diminished response to PHA. All patients except patient 4 had lymphocyte proliferation in response to
Candidastimulation. Lymphocyte proliferation is expressed as patient absolute counts per minute as well as a percentage of the counts per minute for controls (Table 2). All of the controls had normal proliferative responses to both PHA andC. albi-cans(data not shown).
T-cell subsets.PBLs from five of the six CMC patients
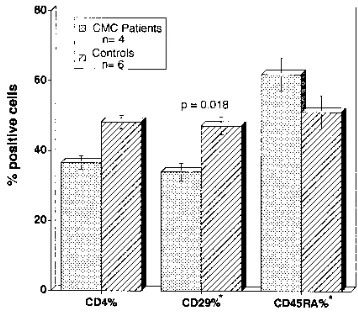
ex-pressed normal absolute numbers of CD41and CD81cells compared with the numbers expressed by age-matched con-trols (Table 2). Patient 4 had had normal CD4 and CD8 cell numbers previously, but experienced a rapid decline in T-cell number and function immediately prior to the present study. CD41/CD291and CD41/CD45RA1cell subsets were quanti-fied for four of the six patients (Fig. 1). The difference in the mean percentage of CD4-positive lymphocytes between pa-tients and controls was not statistically significant (36.5 618 versus 48.3 6 4.6). Patients with CMC had a significant de-crease in the percentage of CD4-positive lymphocytes which coexpressed CD29 compared with the percentage for controls (3466.5 versus 47.266.1;P50.018). CMC patients tended to have a higher percentage of CD4-positive lymphocytes which coexpressed CD45RA than controls (62.067.5 versus 51.3611.8;P50.199), but the difference was not significant.
Cytokine production.The amount of cytokine production by
unstimulated lymphocytes was not significant for either patient or control cells.C. albicans antigen was a poor stimulus for cytokine production by cells from both patients and controls. Lymphocytes from one patient and one control produced de-tectable amounts of IFN-gin response toC. albicans antigen (258.3 and 930 pg/ml, respectively) (Fig. 2). Cells from all patients and controls produced small amounts of IL-4 in
re-FIG. 1. CD4, CD41/CD291, and CD41/CD45RA1cell populations were measured by flow cytometry for six controls and four patients. Results are expressed as percent positive cells. Patients and controls expressed similar amounts of CD4 on their lymphocytes. CMC patients had significantly less CD4-positive lymphocytes which coexpress CD29 than controls (3466.5 versus 47.266.1;P50.018). Patients also tended to have more CD4-positive lympho-cytes which coexpress CD45RA, but the difference was not significant.p, percent CD4-positive cells which coexpress CD29 or CD45RA.
TABLE 2. Lymphocyte proliferation and CD4 and CD8 counts in CMC patients
Patient no.
Lymphocyte proliferationa Total lymphocyte
count (cells/mm3)
Absolute CD4 count (cells/mm3)
Absolute CD8 count (cells/mm3) PHA C. albicans
1 20,729 (106) 11,915 (163) 2,697 971 106 2 33,492 (79) 9,478 (71) 2,352 1,140 799 3 17,489 (122) 18,203 (88) 1,755 788 548
4 4,583 (36) 2,730 (14) 648 36 106
5 61,105 (74) 5,966 (39) 2,304 1,202 507 6 36,817 (69) 7,657 (57) 4,350 1,566 1,131
aData are expressed as absolute counts per minute (percentage of control
counts per minute).
on August 17, 2020 by guest
http://cvi.asm.org/
sponse to C. albicans, but this was not different from the amount produced by unstimulated lymphocytes (range, 18 to 38 pg/ml). PHA-stimulated lymphocytes from patients with CMC did produce significantly more IL-4 in response to PHA than did controls (167 versus 75 pg/ml;P50.001) (Fig. 3). The mean level of production of IFN-gafter stimulation with PHA was not different between controls and patients (455 versus 433 pg/ml). Control lymphocytes produced more IL-2R in re-sponse to PHA than patient lymphocytes; however, the differ-ence was not significant (645 versus 331 pg/ml; P 5 0.17). Control lymphocytes also tended to produce slightly more IL-10 than patient lymphocytes after stimulation with PHA, but again, the difference was not significant (630 versus 402 pg/ml;P50.47).
DISCUSSION
Patients with CMC typically have maintained the ability to produce specific antibodies in response to a variety of antigens includingC. albicans. However, they generally exhibit impaired delayed-type hypersensitivity responses to C. albicans and other antigens. These findings, along with the occurrence of autoimmune disease in patients with CMC, implicate a defect in T-cell regulatory mechanisms in the pathogenesis of the
disease. In the present investigation the patients with CMC had lower levels of circulating CD41/CD291(so-called mem-ory or T-helper inducer cells) cells in their peripheral blood compared with the numbers in healthy controls. These findings are similar to observations in patients with XLA, who also demonstrate cutaneous anergy and poor in vitro responses to recall antigens and who have decreased numbers of circulating CD41/CD291 cells (9). One possible interpretation of the decrease in the number of CD29 cells in patients with XLA is that it is a consequence of repeated immunoglobulin infusions. However, none of our patients with CMC were receiving reg-ular immunoglobulin infusions, so we cannot attribute this observation to the suppression of CD29 cell expression by exogenous immunoglobulin.
A second possible explanation for this phenomenon may be the depletion of CD29 cells from the circulation. CD41/ CD45RA2(memory T) cells accumulate preferentially in in-flamed tissues in association with ELAM-1 (30). Only one of the patients studied (patient 4) demonstrated evidence of ac-tive infection at the time of study, and his CD41/CD291counts were only slightly decreased compared with those of the other patients. None of the other five patients had any evidence of active infection at the time of the study, although patients 1 and 3 had active, ongoing autoimmune diseases. Therefore, it is unlikely that the decrease in the circulating CD29 cell subset is due to an accumulation of these cells in peripheral tissues. A third explanation is that a decrease in the number of circulating CD29 cells is due to a defect in the maturation or activation of CD41/CD45RA1 cells. In vitro, activation of CD4 cells with PHA leads to activation of CD41/CD45RA1T cells, resulting in the downregulation of CD45RA expression and an increase in the level of expression of CD29 and CD45Ro cells, which is associated with the development of T-helper cell function (28). If there is impairment in the acti-vation of CD45RA cells, we would expect to see an increase in this cell population relative to the numbers of cells in the CD29 cell subset. In our study the patients did show a concomitant increase in the CD41/CD45RA1(naive or T-helper suppres-sor) cell population. Therefore, we favor this explanation for the decrease in the level of CD29 expression seen in CMC patients.
We further hypothesize that a decrease in the number of CD29 cells could explain the poor proliferative response in vitro to certain antigens seen in some CMC patients. This would be due to a selective impairment in the activation of CD45RA cells by specific antigens, resulting in a paucity of CD29 cells capable of mounting effector memory responses to these antigens. Moreover, this may also account for the cuta-neous anergy toC. albicans and other antigens because of a predominance of T-suppressor inducer cell activity.
We have also shown that lymphocytes from patients with CMC produce more IL-4 than lymphocytes from controls in response to PHA. IL-4 is produced by TH2 cells and appears to
promote B-cell proliferation and the production of immuno-globulin. IL-4 also downregulates TH1 cells, decreases their
level of production of IL-2, and blocks IL-2-dependent prolif-eration of TH1 cells. Thus, it effectively suppresses the
cell-mediated immune responses cell-mediated by TH1 cells (26). In
some disease states such as leishmaniasis and leprosy, the chronic and destructive disease forms are associated with an impairment in cell-mediated immune responses. These pa-tients show a predominance of IL-4, IL-5, and IL-10 in their lesions, in contrast to the situation in patients with a milder, limited infection, in whom IL-2 and IFN-gpredominate (23, 26). Some studies have suggested that there is a subset of CD8 T cells in mice which, once activated by IL-4, become CD82/ FIG. 2. PBLs (53105) from five patients with CMC and six adult controls
were cultured with and without C. albicansfor 48 h. The supernatant was collected and the amounts of IL-2R, IL-4, IL-10, and IFN-gin the supernatant were measured by ELISA. Neither patients nor controls produced significant amounts of these cytokines from unstimulated cells. There also was no significant production of cytokines byC. albicans-stimulated lymphocytes.p, 0 pg/ml.
FIG. 3. PBLs (53105) from five patients with CMC and six adult controls were also cultured with PHA for 48 to 72 h, and the supernatants were collected. The amounts of IL-2R, IL-4, IL-10, and IFN-gwere measured by ELISA. Lymphocytes from patients with CMC produced significantly more IL-4 in re-sponse to PHA than controls (167 versus 75 pg/ml;P50.001). Patients’ lym-phocytes also tended to produce less IL-2R and IL-10 than control lymlym-phocytes (Pwas not significant).
on August 17, 2020 by guest
http://cvi.asm.org/
CD42and are no longer cytotoxic (12). In patients with leprosy there also appear to be two CD81subsets: the CD81cytotoxic cells which do not produce IL-4 but which do produce IFN-g and IL-10 and CD81T-suppressor cells which produce large amounts of IL-4 (26). Therefore, an increase in the level of IL-4 production in our patients would be expected to result in impairment of cell-mediated immune responses and cytotoxic CD8 cell responses to infection.
In the healthy hostC. albicansmay cause superficial infec-tions but rarely causes severe or disseminated infection. The candidal organism is most likely ingested by macrophages in the skin, andC. albicansantigens are presented with class II major histocompatibility complex molecules to the T-cell re-ceptor on CD4 cells. In the healthy host this probably triggers a cell-mediated immune response, with upregulation and pro-liferation of TH1 cells through the production of IL-2 and
IFN-g. IL-2 production serves to activate CD8 cytotoxic T cells and further control the infection. In animal models in which humoral immunity is suppressed, cell-mediated immunity and innate defenses continue to protect againstC. albicans infec-tion. However, T-cell and cell-mediated immune responses alone did not provide protection against an intravenous chal-lenge, suggesting that antibody production is also important (17). Thus, local host defenses consist primarily of cell-medi-ated immune and cytotoxic T-cell responses. In patients with CMC, we propose that defective regulation of this cell-medi-ated immune response to theC. albicansantigen is due to an inappropriate release of IL-4, which impairs these immune responses and leads to chronic superficial candidal infections. The mechanism of activation of CD45RA cells by which they express CD29 and CD45Ro determinants has not been fully elucidated. When CD45RA cells are activated with PHA in vitro, they express CD29 and show increased levels of expres-sion of CD2, LFA-3, and LFA-1, with decreased levels of expression of CD45RA. These activated cells secrete IFN-g and IL-3 and are more responsive to CD2 and CD3 receptor-mediated activation (1, 27, 28). Both CD45RA and CD29 cells produce IL-2 in response to polyclonal stimulation. However, CD45RA cells produce little or no IL-4 and IFN-g; thus, they can proliferate in response to polyclonal stimulation but pro-vide little helper activity (4). It is not clear whether IL-4 can actually activate CD45RA cells and cause upregulation of CD29 expression, but exposure of T-helper cells to IL-4 does promote the development of short-term effectors with a TH2
phenotype (31, 32). It would appear that even naive CD45RA effector cells are capable of producing TH2 lymphokines.
Therefore, there is no clear marker which can differentiate TH1 and TH2 cell lines; rather, we have markers which reflect
a developmental stage from which T cells may be further dif-ferentiated by their pattern of lymphokine secretion and the type of immune response generated.
In patients with CMC, our findings of a decrease in the numbers of CD41/CD291cells, along with an increase in the level of IL-4 production, further supports the concept that T-helper cell dysfunction contributes to the immunopathogen-esis of CMC. These findings were consistent across all of our study patients, despite their different clinical manifestations. Therefore, these abnormalities alone cannot explain the vari-ous defects seen in CMC; rather, these defects either cause or are part of a cascade of effects which results in various disease processes.
REFERENCES
1.Akbar, A. N., L. Terry, A. Timms, P. C. Beverley, and G. Janossy.1988. Loss of CD45R and gain of UCHL1 reactivity is a feature of primed T cells. J. Immunol.7:2171–2178.
2.Ammann, A. J., and R. Hong.1989. Disorders of the T cell system, p.
257–315.InE. R. Stiehm (ed.), Immunologic disorders in infants and chil-dren, 3rd ed. The W. B. Saunders Co., Philadelphia.
3.Becker, H., A. Langrock, and K. Federlin.1992. Imbalance of CD41 lym-phocyte subsets in patients with mixed connective tissue disease. Clin. Exp. Immunol.88:91–95.
4.Bettens, F., C. Walker, J. Gauchat, D. Gauchat, T. Wyss, and W. Pichler.
1989. Lymphokine gene expression related to CD4 T cell subset (CD45R/ CDw29) phenotype conversion. Eur. J. Immunol.19:1569–1574.
5.Byrne, J., J. Butler, and M. Cooper.1988. Differential activation require-ments for virgin and memory T cells. J. Immunol.141:3249–3257. 6.Chilgren, R. A., P. G. Quie, H. J. Meuwissen, R. A. Good, and R. Hong.1969.
The cellular immune defect in chronic mucocutaneous candidiasis. Lancet
i:1286–1288.
7.Clerici, M., et al.1993. Changes in interleukin-2 and interleukin-4 produc-tion in asymptomatic human immunodeficiency virus-seropositive individu-als. J. Clin. Invest.91:759–765.
8.Clerici, M., et al.1994. Role of interleukin-10 in T helper cell dysfunction in asymptomatic individuals infected with the human immunodeficiency virus. J. Clin. Invest.93:768–775.
9.Crockard, A., N. Boyd, T. McNeill, and D. McCluskey.1992. CD4 lympho-cyte subset abnormalities associated with impaired delayed cutaneous hy-persensitivity reactions in patients with X-linked agammaglobulinemia. Clin. Exp. Immunol.88:29–34.
10. Durandy, A. A., A. Fischer, F. LeDeist, E. Drouchet, and C. Griscelli.1987. Mannan-specific and mannan-induced T-cell suppressive activity in patients with chronic mucocutaneous candidiasis. J. Clin. Immunol.7:400–409. 11. Dwyer, J.1981. Chronic mucocutaneous candidiasis. Annu. Rev. Med.32:
491–497.
12. Erard, F., M. Wild, J. Garcia-Sanz, and G. Le Gros.1993. Switch of CD8 T cells to noncytolytic CD82CD42cells that make TH2 cytokines and help B cells. Science260:1802–1805.
13. Flynn, P. M., F. F. Barrett, and H. G. Herrod.1987. Disseminated histoplas-mosis in two patients with chronic mucocutaneous candidiasis. Pediatr. In-fect. Dis.6:691–692.
14. Herrod, H.1990. Chronic mucocutaneous candidiasis in childhood and com-plications of non-candida infection: a report of the pediatric immunodefi-ciency collaborative study group. J. Pediatr.116:377–382.
15. Kauffman, C. A., M. J. Shea, and P. T. Franne.1981. Invasive fungal infec-tions in patients with chronic mucocutaneous candidiasis. Arch. Intern. Med.
141:1076–1078.
16. Kirkpatrick, C. H.1989. Chronic mucocutaneous candidiasis. Eur. J. Clin. Microbiol. Infect. Dis.8:448–456.
17. Kuruganti, U., L. A. Henderson, R. E. Garner, R. Asofsky, P. J. Baker, and J. E. Domer.1988. Nonspecific and candida-specific immune responses in mice suppressed by chronic administration of anti-m. J. Leukocyte Biol.
44:422–433.
18. Lebranchu, Y., G. Thibault, D. Degenne, and P. Bardos.1990. Deficiency of CD41CD45R1 T lymphocytes in common variable immunodeficiency. N. Engl. J. Med.323:276–277.
19. Matsuyama, T., A. Yamada, D. Rothstein, K. Anderson, S. Schlossman, and C. Morimoto.1991. CD45 isoforms associated with distinct functions of CD4 cells derived from unusual healthy donors lacking CD45RA2T lymphocytes. Cell. Immunol.137:406–419.
20. Morimoto, C., et al.1985. The isolation and characterization of the human helper inducer T cell subset. J. Immunol.134:3762–3768.
21. Oyefara, B., H. Kim, R. Danziger, M. Carroll, J. Greene, and S. D. Douglas.
1994. Autoimmune hemolytic anemia in chronic mucocutaneous candidiasis. Clin. Diagn. Lab. Immunol.1:38–43.
22. Paganelli, R., et al.1992. Selective deficiency of CD41/CD45RA1 lympho-cytes in patients with ataxia-telangiectasia. J. Clin. Immunol.12:84–91. 23. Pirmez, C., M. Yamamura, K. Uyemura, M. Paes-Oliveira, and F.
Concei-cao-Silva.1993. Cytokine patterns in the pathogenesis of human leishma-niasis. J. Clin. Invest.91:1390–1395.
24. Porrini, A., D. Gambi, and G. Malatesta.1992. Memory and naive CD41 lymphocytes in multiple sclerosis. J. Neurol.239:437–440.
25. Richards, S., C. Scott, J. Cole, and H. Gooi.1992. Abnormal CD45R ex-pression in patients with common variable immunodeficiency and X-linked agammaglobulinemia. Br. J. Hematol.81:160–166.
26. Salgame, P., et al.1991. Differing lymphokine profiles of functional subsets of human CD4 and CD8 T cell clones. Science254:279–282.
27. Sanders, M. E., et al.1988. Human memory T lymphocytes express increased levels of three cell adhesion molecules (LFA-3, CD2 and LFA-1) and three other molecules (UCHL1, CDw29 and Pgp-1) and have enhanced IFNg
production. J. Immunol.5:1401–1407.
28. Sanders, M. E., M. W. Makgoba, and S. Shaw.1988. Human naive and memory T cells: reinterpretation of helper-inducer and suppressor-inducer subsets. Immunol. Today9:195–199.
29. Schauer, U., T. Jung, J. Heymanns, and C. Reiger.1991. Imbalance of CD41CD45R1and CD41CD291T helper subsets in patients with atopic dermatitis. Clin. Exp. Immunol.83:25–29.
30. Shimizu, Y., et al.1991. Activation-independent binding of human memory
on August 17, 2020 by guest
http://cvi.asm.org/
T cells to adhesion molecule ELAM-1. Nature (London)349:799–802. 31. Swain, S. L., et al.1991. Helper T-cell subsets: phenotype, function and the
role of lymphokines in regulating their development. Immunol. Rev.123:
115–143.
32. Swain, S. L., G. Huston, S. Tonkonogy, and A. Weinberg.1991. Transforming growth factor-band IL-4 cause helper T cell precursors to develop into
distinct effector helper cells that differ in lymphokine secretion pattern and cell surface phenotype. J. Immunol.147:2991–3000.
33. Twomey, J. T., C. C. Waddell, S. S. Krantz, R. O’Reilly, P. L’Esperance, and R. A. Good.1975. Chronic mucocutaneous candidiasis with macrophage dysfunction, a plasma inhibitor and co-existent aplastic anemia. J. Lab. Clin. Med.85:968–977.