Clay Minerals (1991) 26, 577-582
N O T E
I D E N T I F I C A T I O N O F G R E E N R U S T I N A N O C H R E S L U D G E The bluish-green compounds designated green rusts are layer-structured hydroxides, isostructural with pyroaurite, but containing ferrous and ferric ions in the brucitic sheet (Allman, 1968; Taylor, 1973; Brindley & Bish, 1976). Synthetic preparations of the compounds react readily with oxygen from the atmosphere, causing a change of both structure and colour. Based on the greenish colours often encountered in wet soils and sediments and the rapidity with which this colour changes on exposure to oxygen, Taylor & McKenzie (1980) argued that green rust may form in natural environments. From studies of the oxidation products of synthetic green rust, Taylor (1980) suggested that the green rusts may be important precursors for different iron oxides in soils, depending on environmental conditions. However, the presence of green rust in a natural environment needs to be demonstrated to substantiate its importance. The high susceptibilty towards oxidation is believed to be the major obstacle for the identification of green rust, and various procedures have been designed and demonstrated to stabilize synthetically prepared green rust at room temperature (Taylor, 1982; Hansen, 1989). Keeping the green rust at temperatures much below room temperature also inhibits the oxidation and permits M6ssbauer spectra to be obtained at these temperatures (Murad & Taylor, 1984, 1986; Cuttler et al., 1990). In this note we report the MOssbauer spectra of the greenish eoloured core of an ochre aggregate (before and after partial oxidation) indicating the presence of green rust formed in a natural soil-like environment.
Samples
At the waterworks at Thorsbro 20 km SW of Copenhagen, the ochre sludge, formed by oxidation of ferrous ions in the groundwater, is long-term deposited in freely-drained, open basins --250 m 2 in size. In addition to iron oxide, the freshly deposited ochre also contains calcite. The p H of the sludge is 8.35. Organic matter is added to the sludge in the form of leaves from deciduous trees growing close to the basins, and from willow-herb vegetation growing on the ochre. No accumulation of organic matter was observed, indicating a rapid turn-over of organic matter. At intervals the sludge is transferred to the basin as a suspension, and the consistency of the sludge varies a 10t depending on the water content. The top layer of the sludge in one of the basins consists of rather firm aggregates with diameters between 5 and 20 cm. On breaking some of these aggregates a greenish coloured core surrounded by a reddish brown rind is observed. The relative extent of the core and rind material and the sharp transition between them are clearly shown in the sectioned ochre aggregate in Fig. 1. The green colour of the core gradually vanished over a period of hours.
An absorber for the M6ssbauer investigation of the greenish core was prepared by quickly cutting a sample of the core and pressing it into a Plexiglas absorber holder, which was quickly closed by a Plexiglas lid. An absorber of partially oxidized core material was prepared by grinding a sample of the green core and exposing it to ambient air overnight. After this treatment the sample was placed in a Plexiglas absorber holder. Both samples were quickly frozen by dropping them into liquid nitrogen. In the following, the two samples are referred to as the green core and the oxidized core.
I 1
5 c r n
FIG. 1. Photograph of a sectioned ochre aggregate obtained after 30 s exposure to ambient air.
M~ssbauer s p e c t r a were o b t a i n e d using a c o n v e n t i o n a l constant acceleration spec- t r o m e t e r using an o~-Fe foil at r o o m t e m p e r a t u r e for calibration and as a reference for isomer shifts.
Results and discussion
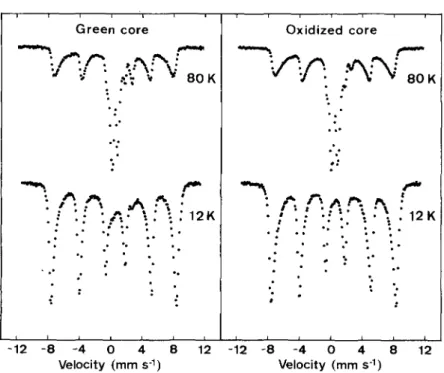
The MOssbauer spectra of the green core and the oxidized core, o b t a i n e d at 80 and 12 K are shown in Fig. 2. B o t h spectra o b t a i n e d at 80 K exhibit a magnetically split sextet with asymmetrically b r o a d e n e d lines, and the central parts of the spectra are d o m i n a t e d by a p a r a m a g n e t i c d o u b l e t due to ferric ions. L o w e r i n g the t e m p e r a t u r e to 12 K caused an increase in the relative a r e a of the magnetically split c o m p o n e n t . H o w e v e r , also at this t e m p e r a t u r e the a b s o r p t i o n lines are asymmetrically b r o a d e n e d . The hyperfine p a r a m e t e r s of the sextet, calculated from the spectra of the green core and the oxidized core at 12 K (Table 1), indicate that goethite is the d o m i n a n t iron oxide in b o t h samples. The hyperfine fields are slightly r e d u c e d as c o m p a r e d to well-crystallized pure goethite. This feature, as well as the asymmetric b r o a d e n i n g of the lines, is most likely caused by p o o r crystallinity ( M u r a d , 1988). T h e t e m p e r a t u r e d e p e n d e n c e o f the s p e c t r a indicate that a m a j o r p a r t of the goethite crystallites is very small and exhibits s u p e r p a r a m a g n e t i c relation at 80 K (M0rup et al., 1980) and t h e r e f o r e contributes to the central ferric doublet. In addition to the sextet, the spectrum of the green core o b t a i n e d at 12 K also exhibits w e a k absorption lines at - 0 - 2 and 2-6 mms 1 indicating the presence of p a r a m a g n e t i c ferrous ions. In the spectra o b t a i n e d at 80 K, the a b s o r p t i o n line at --2-6 mms -1 is m o r e intense and the low-velocity line of this ferrous c o m p o n e n t coincides with the ferric doublet contributing to the a s y m m e t r y of this d o u b l e t . The increased relative a r e a of the F e ( I I ) d o u b l e t at 80 K suggests that p a r t o f the F e ( I I ) c o m p o n e n t is magnetically split at 12 K. T h e spectrum o f the oxidized core at 80 K exhibits only a w e a k a b s o r p t i o n line at - 2 - 6 mms 1 and the ferric d o u b l e t is almost symmetric. In o r d e r to o b t a i n a b e t t e r spectral resolution of the central p a r t of the spectra at 80 K, these spectra were also r e c o r d e d in a smaller velocity range (Fig. 3). A
i t i i J G r e e n c o r e 9 :" "
..','~*"~
~ 8 0 K : 9 t ~ : : i ~ . ~ 2 : : 1 2 K : 9 9 o 9 ~ 1 4 9 i r O x i d i z e d c o r e-r
I 9 , o , 9 o g " f , / ' ~ . t~. ~ 9 .~ - : : . ~ "I, . 1 2 K 9 l ~ 9 ~ 9 . o 9 ~ 9 * 9 # g - 1 2 - 8 - 0 4 1 9 - 1 2 - - 0 4 8 1 2Velocity (mm s -1)
Velocity (rnm s q)
Fl~. 2. M6ssbaner spectra of the green core and the oxidized core obtained at 80 and 12 K at source velocities up to _+12 mms -1.
TABLE 1. M6ssbauer parameters for the sextet at 12 K. For comparison the parameters for well-crystallized goethite are also given.
Bhf (T) e(mms 1) 6(mms-1) FI,6 (mms-t)
Green core 49.8 -0.13 0-49 -0.8
Oxidized core 49.6 -0.11 0-49 -0.9
Well-crystallized
goethite a 50.6 -0.12 0-48 -0.25
B h f = magnetic hyperfine field, e = quadrupole shift, di = isomer shift,
FI,6 = full width at half height of lines 1 and 6.
a Mcirup et al. (1983).
c o m p a r i s o n of t h e two spectra shows t h a t following o x i d a t i o n t h e a b s o r p t i o n line d u e to f e r r o u s i r o n at - 2 . 6 m m s 1 decreases in i n t e n s i t y , a n d is b r o a d e n e d a n d shifted slightly t o w a r d s s m a l l e r velocities. M o r e o v e r , t h e r e s o l u t i o n b e t w e e n the a b s o r p t i o n line at ~ 1 . 0 rams -1 a n d t h e f o u r t h line of t h e sextet at 1.7 rams -1 is r e d u c e d . This i n d i c a t e s t h a t t h e ferric ions, f o r m e d b y o x i d a t i o n of t h e F e ( I I ) ions, c o n t r i b u t e to a p a r a m a g n e t i c c o m p o n e n t with the high-velocity line b e t w e e n 1.0 m m s a a n d 1.7 m m s 1.
If g r e e n rust is f o r m e d in t h e c a r b o n a t e - c o n t a i n i n g o c h r e sludge, it is e x p e c t e d to b e a c a r b o n a t e f o r m , a n d t h e r e f o r e a c o m p a r i s o n with s y n t h e t i c h y d r o x y c a r b o n a t e g r e e n rusts is r e l e v a n t . M u r a d & T a y l o r (1984, 1986) h a v e s t u d i e d t h e M 6 s s b a u e r spectra o f s y n t h e t i c
l 1 1 I
G r e e n
c o r e
"~
e 9 9 . ".g lidO x i d i z e d
c o r e
9
edJb
e 9 i , ]- 2 . 0
0 . 0
.r
I- 6 . 0
- 4 . 0
2.0
4.0
6.0
Velocity (mm s -1)
Fro. 3. M 6 s s b a u e r s p e c t r a o f the g r e e n core a n d oxidized core o b t a i n e d at 80 K at source velocities up
to • rams 1. The line at 2.64 rams 1 is a guide for the change in line position.
hydroxycarbonate green rusts and their oxidation products at 120 K and have demonstrated how readily they react with oxygen. The changes in the M6ssbauer spectra u p o n oxidation were studied for two samples of different composition which showed different behaviour. One sample changed in such a way that the spectrum developed new paramagnetic ferric and ferrous components, whereas the other also developed a magnetically split sextet. These components are due to partially oxidized green rust and iron oxides. For both samples the oxidation resulted in a decrease in the intensity of the ferrous doublets (the spectra were decomposed into two ferric and two ferrous components) and a broadening of the high-velocity line of the ferric components. One of the samples showed a significant shift of the high-velocity line for the ferrous doublet towards smaller velocities. Furthermore, for both samples an increase in the magnetically split part of the low-temperature (liquid helium) spectra following oxidation was reported. It was also found that part of the Fe(II) c o m p o n e n t became magnetically split at this temperature. The changes in the spectra with temperature and upon partial oxidation are qualitatively identical to those seen in the spectra of the present samples. At 80 K the superparamagnetic ferric doublet overlaps the
contributions of the paramagnetic ferric and ferrous components of the green core making it difficult to obtain reliable M0ssbauer parameters of the individual components. However, the position of the well-resolved ferrous line at --2-6 mms - I may be used to compare with results obtained from studies of synthetic samples. For the sample form the green core, this line is positioned at 2.64 m m s - t (at 80 K), which may be compared to line positions of 2.55- 2.60 mms t (at 120 K) for the most intense ferrous doublet as calculated from the data of Murad & Taylor (1984, 1986). Although these line positions are in good agreement, the line-width of the ferrous component in the green core sample is much higher than that of the most intense c o m p o n e n t in the synthetic samples (0.47 mms 1 and 0-28 mms -1, respectively). Inasmuch as the sample from the green core is a natural sample, its crystal chemistry may well be more complex (including partial oxidation in its natural state) leading to broadening of the lines. Thus the results indicate that the ferrous c o m p o n e n t found in the green core is due to green rust. The observation that the Fe(II) c o m p o n e n t becomes partially magnetically split at 12 K is also in accordance with this conclusion.
A n X-ray powder diffractogram of the oxidized core revealed only calcite and iron oxides (goethite is dominant, but small amounts of ferrihydrite can not be excluded), indicating that the mineralogy is relatively simple. Samples from other natural environments containing green rust, may, for example, also contain ferrous layer-silicates which may have components with absorption lines at - 2 . 6 mms 1 and which oxidize relatively easily. The presence of such components makes identification of green rust very difficult. However, in contrast to the ferric oxides, which are the final oxidation products of green rusts, most ferric components in layer-silicates are expected to remain paramagnetic at temperatures above 12 K. A n o t h e r important factor facilitating detection of green rust in the present sample is its relatively high content. From the 80 K spectrum of the green core it is estimated that ferrous iron amounts to - 6 % of the total iron, and assuming a ferrous to ferric ratio of 1-5 in the green rust, it is estimated that - 10% of the Fe in the sample is present in the green rust. The morphology of the green core, the sharp colour transition, and the absence of ferrous iron in the freshly precipitated ochre indicate that anaerobic transformation of organic matter plays a major role in the reduction of ferric compounds necessary for the formation of green rust.
In conclusion, the results presented indicate that a green rust c o m p o u n d had formed by natural processes in the ochre sludge.
Laboratory of Applied Physics, Technical University of D e n m a r k , DK-2800 Lyngby,
Denmark.
Received 11 March 1991; revised 8 April 1991.
C. BENDER KOCH S. MORUP
REFERENCES
ALLMANN R. (1968) The crystal structure of pyroaurite. Acta Cryst. B24, 972-977.
BRINDLEY G.W. & BISH D.L. (1976) Green rust: a pyroaurite type structure. Nature 263, 353.
CU~LER A,H., MAN V., CRAr~SnAW T.E. & LONOWORTH G. (1990). A M6ssbauer study of green rust precipitates: I.
Preparation from sulphate solutions. Clay Miner. 25, 289-30l.
HANSeN H.C.B. (1989) Composition, stabilization, and light absorption of Fe(II)Fe(III) bydroxycarbonate ("green rust"). Clay Miner. 24, 663-669.
BREaN C., DEAN~ A+T. & FLYNN J.J. (1987) The acidity of trivatent cation-exchanged montmorillonite+
Temperature programmed desorption and infrared studies of pyridine and n-butylamine. Clay Miner. 22,
169-178.
BREEN C. (1991) Thermogravimetric study of the desorption of cyclohexylamine and pyridine from an acid-treated
Wyoming bentonite. Clay Miner. 26, 473M86.
CALVE'r R. & PROST R. (1971) Cation migration into empty octahedral sites and surface properties of clays, Clays
Clay Miner. 19, 175-186.
DEANE A.T, (1987) Physicochemical studies of clay-organic interactions. PhD thesis, NIHE, Dublin, Eire.
GASSER R.P.H. (1987) An Introduction to Chemisorption and Catalysis by Metals, pp. 66-71. Oxford Science
Publications, Clarendon Press, Oxford.
HOFMANN V. & KLEMEN R. (1950) Verlust der Austaush fahigkeit yon Lithiumionen und Bentonit durch Erhitzung. Z. Anorg. Allg. Chem. 262, 95-99.
KOPPELMAN M.H. & DILLARD J.G. (1977) A study of the adsorption of Ni(II) and Cu(ll) by clay minerals. Clays
Clay Miner. 25, 457M62.
THOMAS J.M. (1982) Sheet silicate intercalates: New agents for unusual chemical conversions. Pp. 56-97 in: Intercalation Chemistry (M.S. Whittingham & A.J. Jacobson, editors). Academic Press, New York. TRONCON! F~. & FORZNrT! P. (1985) Experimental criteria for diffusional limitations during temperature
programmed desorption from porous catalysts. J. Catal. 93, 197-200.
VELGHE F., SCHOONHEYDr R.A. & UYTTERHOEVEN J.B. (1977) The co-ordination of hydrated Cu(II)- and Ni(II)-ions
on montmorillonite surface. Clays Clay Miner. 25, 375-380.
WARD J.W. (1968) Spectroscopic study of the surface of zeolite Y: the adsorption of pyridine. J. Coll. lnter. Sci. 28,