0095-1137/10/$12.00 doi:10.1128/JCM.00542-09
Copyright © 2010, American Society for Microbiology. All Rights Reserved.
Diversity of
Staphylococcus
Species Strains Based on Partial
kat
(Catalase)
Gene Sequences and Design of a PCR-Restriction Fragment Length
Polymorphism Assay for Identification and Differentiation of
Coagulase-Positive Species (S. aureus,
S. delphini,
S. hyicus,
S. intermedius,
S. pseudintermedius, and
S. schleiferi
subsp.
coagulans)
䌤
Giuseppe Blaiotta,
1* Vincenzina Fusco,
3Danilo Ercolini,
2Olimpia Pepe,
1and Salvatore Coppola
1 Department of Food Science, School of Agriculture,1and School of Biotechnological Sciences,2University of Naples Federico II,Via Universita` 100, 80055 Portici, Italy, and National Research Council, Institute of Sciences of Food Production, Via Amendola 122/o, 70126 Bari, Italy3
Received 17 March 2009/Returned for modification 22 June 2009/Accepted 23 October 2009
A set of degenerate PCR primers was designed and used to amplify and sequence about 75% of the catalase (kat) gene from each of 49 staphylococcal strains. In some strains ofStaphylococcus xylosus,S. saprophyticus, and
S. equorum, two catalase genes, katA andkatB, were found. A phylogenetic tree was generated and showed
diversities among 66 partial (about 900-bp) staphylococcalkatnucleotide sequences (including 17 sequences found in GenBank) representing 26 different species. The topology of this tree showed a distribution of staphylococcal species similar, but not identical, to those reported previously based on 16S rRNA,hsp60,sodA,
rpoB,tuf, andgapgenes. Thekatgene sequences were less conserved than those of 16S rRNA,rpoB,hsp60, and
tufgenes and slightly more conserved than those of thegapgene. Therefore,katgene sequence analysis may provide an additional marker for inferring phylogenetic relationships of staphylococci. Moreover, the discrete nucleotide polymorphism revealed in this gene could be exploited for rapid, low-cost identification of staph-ylococcal species through PCR-restriction fragment length polymorphism (RFLP) analysis. In this study, a PCR-RFLP assay performed by using only the TaqI restriction enzyme was successfully developed for rapid unequivocal identification/differentiation, at species and subspecies levels, of coagulase-positive staphylococci (CPS). The assay was validated by testing the DNA from 100 staphylococcal strains, including reference and wild CPS strains isolated from different environments. This reliable, rapid, and low-cost approach (requiring about 6 h from DNA isolation to the achievement of results and <5 Euros for each strain tested) allowed unambiguous identification of all the strains assayed, including the newly describedS. delphiniandS.
pseud-intermediusCPS species.
Staphylococci are a group of microorganisms occurring widely in nature. As reported in the List of Prokaryotic Names with Standing in Nomenclature (www.bacterio.net) as of the full update in March 2006, theStaphylococcusgenus includes 39 valid species, 11 of which are divided into two or more subspecies, resulting in more than 50 recognized systematic entities (15).
The genus includes both human and animal pathogens, gen-erally coagulase-positive staphylococci (CPS) such asS. aureus,
S. intermedius,S. delphini,S. hyicus,S. schleiferisubsp. coagu-lans, andS. pseudintermedius(12, 21, 29, 35), and coagulase-negative staphylococci (CNS) such asS. equorum,S. xylosus,S. carnosus,S. simulans,S. saprophyticus,S. succinus,S. warneri,
S. vitulinus,S. pasteuri,S. epidermidis, andS. lentus.
Notwithstanding their valuable role in food fermentation (5–9, 23, 24), some CNS such asS. saprophyticus,S. epidermi-dis, andS. haemolyticusexhibit increasing abilities as
opportu-nistic/emerging pathogens in colonizing animal and human tissues, due in part to the ability of these species (particularly
S. epidermidis) to form protective biofilms and their ubiquitous occurrence in the environment (37). Finally, the occurrence of some pathogenic species (S. aureus,S. intermedius, andS. hyi-cus) in food is a potential public health hazard, since many strains can produce enterotoxins (4, 9, 31). Indeed, entero-toxin-producing food-associated CNS strains (S. carnosus, S. equorum,S. piscifermentans, andS. xylosus) were also recently described by Zell et al. (40). Despite the importance of accu-rate identification of staphylococcal species by microbiological laboratories, previous studies demonstrated the unreliability of phenotypic methods, including the Vitek 2 system (bio-Me´rieux, Marcy l’Etoile, France), compared to different mo-lecular techniques (5–8, 11) to identify staphylococci.
Due to the high sensitivity and specificity they provide, mo-lecular markers are an alternative tool for accurate identifica-tion and classificaidentifica-tion ofStaphylococcusspecies. Evaluations of 16S to 23S rRNA gene polymorphisms by PCR and PCR-denaturing gradient gel electrophoresis (DGGE) analyses of 16S ribosomal sequences have been assessed for the ability to achieve clear identification of strains within theStaphylococcus
* Corresponding author. Mailing address: Dipartimento di Scienza degli Alimenti, Universita` degli Studi di Napoli Federico II, Via Uni-versita`, 100, 80055 Portici (Naples), Italy. Phone: 39-081-2539451. Fax: 39-081-2539407. E-mail: [email protected].
䌤Published ahead of print on 4 November 2009.
192
on May 16, 2020 by guest
http://jcm.asm.org/
genus (6). Molecular assays targeting some housekeeping genes such ashsp60(17), the 16S rRNA gene (14, 18),femA
(36),tuf(22),gap(38, 39),sodA(26),rpoB(13), anddnaJ(20, 30) have been used for reliably identifying and classifying staphylococci. However, except for thesodA,hsp60, andnuc
gene sequence analyses carried out by Sasaki et al. (29), which allowed some phenotypically identifiedS. intermediusstrains to be reclassified asS. delphini and S. pseudintermedius, to our knowledge, no method allowing rapid identification and differ-entiation of CPS species including the recently described S. delphiniandS. pseudintermediusis available to date.
In this study, we evaluated the catalase (kat) gene as a new target for phylogenetic analysis of staphylococci. Catalase is a heme-containing enzyme involved in dismutation of hydrogen peroxide generated during cellular metabolism to water and molecular oxygen. InS. aureus, a correlation between catalase activity and virulence has been observed, suggesting a role for catalase in defensive mechanisms against the oxygen radicals produced by macrophages (28). Additionally, a recent study (25) showed that theS. aureuscatalase is a major factor inS. aureusdefense againstStreptococcus pneumoniaedue to neu-tralization of secreted H2O2 produced by the latter microor-ganism. Sanz et al. (28) showed that catalase deficiency inS. aureussubsp.anaerobiusis associated with natural loss-of-func-tion mutaloss-of-func-tions within the structural gene. Therefore, catalase-negative staphylococcal strains may also harbor the catalase gene. Barrie`re et al. (3) described thekatAgene ofS. xylosus
C2a and supposed the presence of a second catalase gene (katB) in this strain. In fact, thekatA-deficient mutant of S. xylosus constructed by these authors still exhibited catalase activity. After analysis of polymorphism within thekatgenes of 26 different staphylococcal species, we designed and success-fully applied a molecular assay allowing rapid and unequivocal identification and differentiation of CPS species and subspecies.
MATERIALS AND METHODS
Bacterial strains.Forty-six reference strains (including type strains and pre-viously identified strains) representing 25 different staphylococcal species were used in this study (Fig. 1). Moreover, to validate the implementedkatA PCR-restriction fragment length polymorphism (RFLP) approach for rapid identifi-cation of CNS, other staphylococcal strains from different environments were also analyzed (Table 1). Working cultures were grown in brain heart infusion broth (Oxoid Ltd., Basingstoke, Hampshire, United Kingdom) at 37°C. Before DNA extraction, cultures were streaked onto brain heart infusion agar plates and grown overnight at 37°C.
DNA isolation.DNA extraction from a single colony was carried out by using the InstaGene matrix under the conditions described by the supplier (Bio-Rad Laboratories, Hercules, CA).
Amplification and sequencing ofkatgenes.Degenerate primers KDN-For (AARGGWTCHGGWGCWTTYGG) and KDN-Rev (TGTTCRAARTARTT RTCRTCATC) were selected from among a highly conserved region of staph-ylococcal strainkatA gene sequences available in the GenBank and EMBL databases (1); the sequences were fromS. saprophyticus,S. warneri,S. epidermi-dis,S. haemolyticus,S. xylosus,S. aureussubsp.anaerobius, andS. aureussubsp.
aureus(Fig. 1). The primers amplify an internal fragment of 1,114 bp of thekatA
gene, from position 164 to position 1277 (nucleotide numbering with respect to thekatAgene ofS. xylosusC2a [accession no. AJ295151]).
PCR amplifications were performed with a total volume of 50l, including 5 to 10l (25 to 50 ng) of target DNA, 5l ofTaqDNA polymerase 10⫻buffer (Invitrogen S.R.L., Milan, Italy), 2.5l of 50 mM MgCl2, 0.5l of
deoxynucleo-side triphosphate (dNTP) mix (25 mM [each] dNTPs), 0.2l of each primer (0.1 mM), and 0.5l of aTaqDNA polymerase solution (5 U/l; Invitrogen S.R.L.). PCR thermal conditions consisted of an initial denaturing step (95°C for 3 min), 40 amplification cycles (60 s at 94°C, 60 s at 52°C, and 90 s at 72°C), and one final
step at 72°C for 10 min. The PCR amplification fragments were resolved by agarose (1.5%, wt/vol) gel electrophoresis at 100 V for 2 h. The gel was stained with ethidium bromide, and the bands were visualized under UV illumination at 254 nm.
FIG. 1. Neighbor-joining tree showing the phylogenetic relation-ships among staphylococci based on a comparison of 900-bpkatgene sequences. Bootstrap values based on 1,000 replications are given at the branching points when they are above 50%. The scale bar evaluates the sequence divergence. EMBL accession numbers are given in pa-rentheses.*,katgene sequence(s) was determined during this study.
on May 16, 2020 by guest
http://jcm.asm.org/
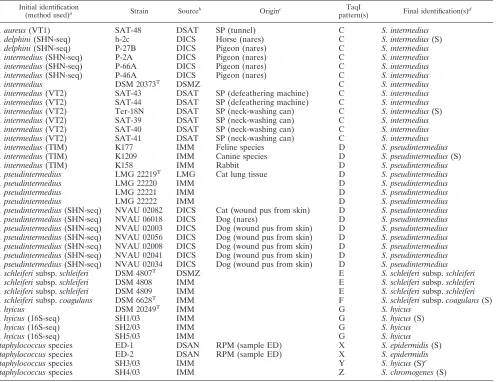
TABLE 1. Sources, origins, TaqI RFLP patterns, and identification of some CPS strains used to validate our RFLP approach
Initial identification
(method used)a Strain Sourceb Originc
TaqI
pattern(s) Final identification(s)
d
S. aureus(sp-PCR) D4508 CNTS A S. aureus
S. aureus NCTC 9393 CNTS A S. aureus
S. aureus ATCC 27664 CNTS Chicken tetrazzini A S. aureus S. aureus ATCC 25923 ATCC Clinical specimen A S. aureus S. aureus ATCC 14458 ATCC Feces from child A S. aureus
S. aureus(sp-PCR) E DSAN FLC A S. aureus
S. aureus(sp-PCR) F DSAN FLC A S. aureus
S. aureus(sp-PCR) G DSAN FLC A S. aureus
S. aureus(sp-PCR) H DSAN FLC A S. aureus
S. aureus(sp-PCR) I DSAN FLC A S. aureus
S. aureus(sp-PCR) M DSAN FLC A S. aureus
S. aureus(sp-PCR) N DSAN FLC A S. aureus
S. aureus(sp-PCR) T(C14) DSAN FLC A S. aureus
S. aureus(sp-PCR) X(CS14) DSAN FLC A S. aureus S. aureus(sp-PCR) K4644/97 IMM HP (blood culture) A S. aureus S. aureus(sp-PCR) K6278/97 IMM HP (blood culture) A S. aureus S. aureus(sp-PCR) A1048/98 IMM HP (nasal swab) A S. aureus S. aureus(sp-PCR) A2586/99 IMM HP (nasal swab) A S. aureus S. aureus(sp-PCR) A2812/98 IMM HP (nasal swab) A S. aureus S. aureus(sp-PCR) A4178/98 IMM HP (nasal swab) A S. aureus S. aureus(sp-PCR) A652/99 IMM HP (nasal swab) A S. aureus S. aureus(sp-PCR) 7645a IMM HP (patient with osteomyelitis) A S. aureus S. aureus(sp-PCR) OM 56/2a IMM HP (patient with osteomyelitis) A S. aureus S. aureus DSM 20231T DSMZ Human pleural fluid A S. aureus S. aureus ATCC 19095 CNTS Leg abscess fluid A S. aureus S. aureus(sp-PCR) SI1 DSAN MCM (plant A) A S. aureus S. aureus(sp-PCR) SI9 DSAN MCM (plant A) A S. aureus S. aureus RIMD 31092 CNTS MRSA strain A S. aureus S. aureus(sp-PCR) AS14g DSAN NTS (sample A) A S. aureus S. aureus(sp-PCR) AS27 DSAN NTS (sample A) A S. aureus S. aureus(sp-PCR) BS4 DSAN NTS (sample B) A S. aureus S. aureus(sp-PCR) DS18g DSAN NTS (sample D) A S. aureus S. aureus A900322 CNTS Patient with TSS A S. aureus S. aureus(sp-PCR) AB-8802 DSAN RPM (sample AB) A S. aureus S. aureus(sp-PCR) ED-3 DSAN RPM (sample ED) A S. aureus S. aureus(sp-PCR) ED-4 DSAN RPM (sample ED) A S. aureus S. aureus 382F AFSSA Unspecified (food) A S. aureus S. aureus(sp-PCR) SMDE DSAN Unspecified (human specimen) A S. aureus S. aureus(sp-PCR) LA14 DSAN WBRM (plant A) A S. aureus S. aureus(sp-PCR) R1 DSAN WBRM (plant A) A S. aureus S. aureussubsp.anaerobius DSM 20714T IMM A S. aureus S. aureus(VT1) SAT-54 DSAT SP (eyebrow of operator) A S. aureus S. aureus(VT1) SAT-56 DSAT SP (eyebrow of operator) A S. aureus S. aureus(VT1) SAT-53 DSAT SP (hand of operator) A S. aureus S. aureus(VT2) SAT-58 DSAT SP (conjunctiva of operator) A S. aureus S. aureus(VT2) SAT-59 DSAT SP (conjunctiva of operator) A S. aureus S. aureus(VT2) Ter-105 DSAT SP (defeathering machine) A S. aureus S. aureus(VT2) Ter-106 DSAT SP (defeathering machine) A S. aureus S. aureus(VT2) Ter-107 DSAT SP (defeathering machine) A S. aureus S. aureus(VT2) Ter-109 DSAT SP (defeathering machine) A S. aureus S. aureus(VT2) SAT-55 DSAT SP (eyebrow of operator) A S. aureus S. aureus(VT2) SAT-57 DSAT SP (eyebrow of operator) A S. aureus S. aureus(VT2) Ter-10 DSAT SP (gutting machine) A S. aureus
S. intermedius(VT2) SAT-42 DSAT SP (neck-washing can) A and C S. aureus/S. intermedius S. aureus(VT1) SAT-51 DSAT SP (apron of operator) B S. delphini
S. aureus(VT1) SAT-52 DSAT SP (apron of operator) B S. delphini S. chromogenes(VT2) Ter-24N DSAT SP (hand of operator) B S. delphini(S) S. delphini LMG 22190T LMG Dolphin B S. delphini S. delphini(SHN-seq) h-9D DICS Horse (nares) B S. delphini(S) S. delphini(SHN-seq) P14A DICS Pigeon (nares) B S. delphini S. intermedius(TIM) K1224 IMM Equine species B S. delphini(S) S. intermedius(VT2) SAT-49 DSAT SP (apron of operator) B S. delphini S. intermedius(VT2) SAT-50 DSAT SP (apron of operator) B S. delphini S. intermedius(VT2) Ter-25N DSAT SP (hand of operator) B S. delphini S. intermedius(VT2) SAT-45 DSAT SP (tunnel) B S. delphini S. intermedius(VT2) SAT-46N DSAT SP (tunnel) B S. delphini S. intermedius(VT2) SAT-47N DSAT SP (tunnel) B S. delphini
Continued on following page
on May 16, 2020 by guest
http://jcm.asm.org/
The PCR products of the expected length were purified from the agarose gel by using a QIAquick gel extraction kit (Qiagen S.p.A., Milan, Italy) and quan-tified by comparison with lambda/HindIII molecular marker fragments (Invitro-gen S.R.L.).
Before sequencing, restriction digestion of the purified PCR products was performed in a total volume of 50l by using about 300 ng of DNA and 30 U of CfoI restriction endonuclease enzyme (Promega Italia S.R.L., Milan, Italy) so as to evaluate the specificity of the amplified fragment and/or the presence of spurious PCR products (see Results).
The DNA sequences were determined by the dideoxy chain termination method by using a DNA sequencing kit (Perkin-Elmer Cetus, Emeryville, CA) and primer KDN-For. The sequences were analyzed by MacDNasis Pro version 3.0.7 (Hitachi Software Engineering Europe S.A., Olivet, France), and research on DNA similarity was performed with the GenBank and EMBL databases (1). Phylogenetic analysis was carried out using MEGA version 4.0 (33) after
multiple-sequence alignment of data by CLUSTAL W 1.8 (34). Distance matrix and neighbor-joining methods (27) were applied for tree construction.
DNA sequence similarity analysis was performed by BioEdit version 7.0.9 (www.mbio.ncsu.edu/BioEdit/bioedit.html).
Design of the PCR-RFLP assay for identification and differentiation ofS. aureus,S. delphini,S. hyicus,S. intermedius,S. pseudintermedius, andS. schleiferi.
From the highly conserved region ofS. aureus,S. delphini,S. hyicus,S. interme-dius, S. pseudintermedius, and S. schleiferi katA gene sequences found in GenBank or determined during this study (Fig. 1), two oligonucleotide primers were selected: CPSK1F (CARAAYAACTGGGATTTCTGGAC) and CPSK6R (GCATCRCCRTAWGAGAATAAACG). Targeting positions 487 to 509 and 1031 to 1009 of thekatAgene ofS. aureussubsp.aureusMu50 (BA000017) allowed amplification of fragments of 544 bp. PCR amplifications were per-formed with a total volume of 50l, including 5l of target DNA, 5l ofTaq
[image:4.585.48.544.81.463.2]DNA polymerase 10⫻buffer (Invitrogen), 2.5l of 50 mM MgCl2, 0.5l of
TABLE 1—Continued
Initial identification
(method used)a Strain Sourceb Originc
TaqI
pattern(s) Final identification(s)
d
S. aureus(VT1) SAT-48 DSAT SP (tunnel) C S. intermedius S. delphini(SHN-seq) h-2c DICS Horse (nares) C S. intermedius(S) S. delphini(SHN-seq) P-27B DICS Pigeon (nares) C S. intermedius S. intermedius(SHN-seq) P-2A DICS Pigeon (nares) C S. intermedius S. intermedius(SHN-seq) P-66A DICS Pigeon (nares) C S. intermedius S. intermedius(SHN-seq) P-46A DICS Pigeon (nares) C S. intermedius
S. intermedius DSM 20373T DSMZ C S. intermedius
S. intermedius(VT2) SAT-43 DSAT SP (defeathering machine) C S. intermedius S. intermedius(VT2) SAT-44 DSAT SP (defeathering machine) C S. intermedius S. intermedius(VT2) Ter-18N DSAT SP (neck-washing can) C S. intermedius(S) S. intermedius(VT2) SAT-39 DSAT SP (neck-washing can) C S. intermedius S. intermedius(VT2) SAT-40 DSAT SP (neck-washing can) C S. intermedius S. intermedius(VT2) SAT-41 DSAT SP (neck-washing can) C S. intermedius S. intermedius(TIM) K177 IMM Feline species D S. pseudintermedius S. intermedius(TIM) K1209 IMM Canine species D S. pseudintermedius(S) S. intermedius(TIM) K158 IMM Rabbit D S. pseudintermedius S. pseudintermedius LMG 22219T LMG Cat lung tissue D S. pseudintermedius S. pseudintermedius LMG 22220 IMM D S. pseudintermedius S. pseudintermedius LMG 22221 IMM D S. pseudintermedius S. pseudintermedius LMG 22222 IMM D S. pseudintermedius S. pseudintermedius(SHN-seq) NVAU 02082 DICS Cat (wound pus from skin) D S. pseudintermedius S. pseudintermedius(SHN-seq) NVAU 06018 DICS Dog (nares) D S. pseudintermedius S. pseudintermedius(SHN-seq) NVAU 02003 DICS Dog (wound pus from skin) D S. pseudintermedius S. pseudintermedius(SHN-seq) NVAU 02056 DICS Dog (wound pus from skin) D S. pseudintermedius S. pseudintermedius(SHN-seq) NVAU 02008 DICS Dog (wound pus from skin) D S. pseudintermedius S. pseudintermedius(SHN-seq) NVAU 02041 DICS Dog (wound pus from skin) D S. pseudintermedius S. pseudintermedius(SHN-seq) NVAU 02034 DICS Dog (wound pus from skin) D S. pseudintermedius S. schleiferisubsp.schleiferi DSM 4807T DSMZ E S. schleiferisubsp.schleiferi S. schleiferisubsp.schleiferi DSM 4808 IMM E S. schleiferisubsp.schleiferi S. schleiferisubsp.schleiferi DSM 4809 IMM E S. schleiferisubsp.schleiferi S. schleiferisubsp.coagulans DSM 6628T IMM F S. schleiferisubsp.coagulans(S)
S. hyicus DSM 20249T IMM G S. hyicus
S. hyicus(16S-seq) SH1/03 IMM G S. hyicus(S)
S. hyicus(16S-seq) SH2/03 IMM G S. hyicus
S. hyicus(16S-seq) SH5/03 IMM G S. hyicus
Staphylococcusspecies ED-1 DSAN RPM (sample ED) X S. epidermidis(S) Staphylococcusspecies ED-2 DSAN RPM (sample ED) X S. epidermidis Staphylococcusspecies SH3/03 IMM Y S. hyicus(S)e
Staphylococcusspecies SH4/03 IMM Z S. chromogenes(S)
a
Identification methods: sp-PCR, species-specific PCR assays targetingnucandhsp60genes (9, 10); VT1 and VT2, testing by Vitek 1 and Vitek 2 systems (bioMérieux, Marcy l’Etoile, France), respectively; SHN-seq, sequencing ofsodA,hsp60, andnucgenes (29); TIM, traditional identification methods (K. Becker, personal communication); and 16S-seq, 16S rRNA gene sequencing (K. Becker, personal communication).
b
CNTS, Centre Nationale des Toxémies a Staphylococques, Faculté de Médecine Laennee, France (kindly provided by G. Lina); ATCC, American Type Culture Collection, Rockville, MD; DSAN, Dipartimento di Scienza degli Alimenti, Universita` degli Studi di Napoli Federico II, Italy; IMM, Institut fu¨r Medizinische Mikrobiologie, Universita¨tsklinikum Mu¨nster, Germany (kindly provided by K. Becker); DSMZ, Deutsche Sammlung von Mikroorganismen und Zellkulturen, Braunschweig, Germany; AFSSA, Agence Franc¸aise de Sécurité Sanitaire des Aliments, France (kindly provided by P. Fach); DSAT, Dipartimento di Scienze degli Alimenti, Universita` degli Studi di Teramo, Italy (kindly provided by A. Ianieri); LMG, Laboratorium voor Microbiologie bacteria collection, Universiteit Gent, Belgium; DICS, Department of Infection Control Science, Juntendo University, Japan (kindly provided by K. Hiramatsu).
c
FLC, Fior di latte cheese; HP, hospital patient; MCM, water buffalo mozzarella cheese manufacturing plant (natural whey cultures); MRSA, methicillin-resistant
S. aureus; NTS, “Napoli-type” salami; TSS, toxic shock syndrome; RPM, raw poultry meat; WBRM, water buffalo raw milk; SP, slaughterhouse for pigeons.
d
For some strains, identified by “(S),” identification was confirmed by partial sequencing of thekatAandgapgenes.
e
Found to have the highest level of similarity, 88%, toS. hyicus.
on May 16, 2020 by guest
http://jcm.asm.org/
dNTP mix (25 mM [each] dNTPs), 0.2l of each primer (0.1 mM), and 0.2l of
TaqDNA polymerase solution (5 U/l; Invitrogen S.R.L.). PCR thermal condi-tions consisted of an initial denaturing step (95°C for 3 min) and 40 amplification cycles: a denaturing step for 10 s at 95°C and an annealing-extension step for 45 s at 56°C. After amplification, 15-l samples of PCR mixtures were tested by agarose (1.5%, wt/vol) gel electrophoresis at 100 V for 1 h. The remaining part (30l) of the PCR product was digested in a total volume of 50l by 20 U of TaqI restriction endonuclease (GE Healthcare, Milan, Italy) at 65°C for 2 h. Restriction fragments were resolved by agarose (2%, wt/vol) gel electrophoresis at 100 V for 2 h.
Nucleotide sequence accession number.The sequence of thekatBgene ofS. xylosusDSM 20266Tdetermined in this study was deposited in GenBank under
accession number AY702101.
RESULTS
Sequencing of the Staphylococcus kat genes. Partial se-quences (⬍900 bp) of the kat genes from 49 staphylococcal strains were determined by using degenerate PCR primers designed during this study. Initially, for only 46 of 49 staphy-lococcal strains available, it was possible to directly sequence more than 900 bp of the purified 1,114-bp KDN-For/KDN-Rev PCR product. Gene sequences obtained fromS. saprophyticus
strains P72K3, P52K3, and GB1,S. warneristrains DSM 20316 and P-98M5,S. epidermidisstrains DSM 1798 and DSM 20044,
S. haemolyticusstrain DSM 20263, andS. aureusstrains ATCC 27664 and DSM 20231 displayed a high level of similarity to
katAgene sequences from strains of the same species reported in the database (Fig. 1), confirming that the amplified products were really part of thekatAgene.
It was not possible to achieve direct sequencing of the purified 1,114-bp KDN-For/KDN-Rev PCR product fromS. xylosusDSM 20266T,S. equorumsubsp. linensDSM 15097T, andS.
saprophyt-icussubsp. saprophyticusDSM 20229Tstrains. In fact, in analyzing
the CfoI KDN-For/KDN-Rev restriction pattern of S. xylosus
DSM 20266T, we observed that in some cases the sum of the
molecular sizes of the restriction fragments was about 2,200 bp, i.e., double the expected length (Fig. 2, pattern C). This result was confirmed when 1g of PCR product was digested with 30 U of CfoI in 16 h at 37°C, excluding the hypothesis of partial digestion of the PCR product. Moreover, 23 of the 31S. xylosus strains analyzed showed restriction profiles similar to that of DSM 20266T (data not shown). The presence of a second katgene
(katB) in strainS. xylosusDSM 20266T, similar to the one
de-scribed by Barrie`re et al. (3), was indicated by TaqI restriction digestion of the KDN-For/KDN-Rev PCR product, and the full nucleotide sequence (accession number AY702101) was deter-mined by inverse PCR experiments, i.e., digesting total DNA with TaqI and using KBSxFI2 (CCTGACGAAGCAGCGAAAAT) and KBSxFI4 (ACGCGTTCACCTTTGTCGTT) as inverse primers in the PCR.
As shown in Fig. 2 by the CfoI KDN-For/KDN-Rev restric-tion patterns forS. equorumsubsp. linensDSM 15097T
(pat-tern B) andS. saprophyticusDSM 20229T(ATCC 15305;
pat-tern F), these strains also produced additional fragments, suggesting again the presence of two catalase genes. By apply-ing the approach described above forS. xylosusDSM 20266T,
it was possible to determine partial sequences of both catalases of S. equorum and S. saprophyticus. The S. equorum subsp.
linens DSM 15097TkatAgene clustered with S. xylosus katA
genes, whilekatBclustered with S. saprophyticus katAandS. xylosus katB genes (Fig. 1). The katA gene sequence of S.
saprophyticusDSM 20229T determined during this study was
identical to that already described forS. saprophyticusATCC 15305 (accession number AP008934), while katB revealed about 98% similarity tokatAgenes ofS. xylosusstrains (Fig. 1).
katgene sequence similarity and phylogeny derived fromkat
sequences.A total of 66 partialStaphylococcus katsequences (including 17 sequences found in the GenBank and EMBL databases [www.ncbi.nlm.nih.gov]) representing 26 different species were compared. The identity matrix (developed by BioEdit) based on comparison of thekatAgene sequences of 30 Staphylococcus strains revealed that the interspecies se-quence similarity ranged from 0.40 to 0.93. The latter value was calculated forS. carnosusDSM 20501 andS. condimentiDSM 11674, as well as forS. pseudintermediusLMG 22219 and S. intermediusDSM 20273. Sequence similarity values of⬎0.96 were found for strains belonging to subspecies of the same species: S. aureus subsp. anaerobius MVF213 and S. aureus
subsp.aureusDSM 20231 (0.985),S. schleiferisubsp.schleiferi
DSM 4807 andS. schleiferisubsp.coagulansDSM 6628 (0.96), andS. succinussubsp.caseiDSM 15096 andS. succinussubsp.
succinusDSM 14617 (0.96).
Evolutionary divergence between the 66katgene sequences was estimated by using MEGA version 4.0 (33). The number of base differences per site obtained by averaging over all se-quence pairs was 0.197 (standard error, 0.01). The dendrogram resulting from the neighbor-joining analysis of 66 kat se-quences is shown in Fig. 1. The topology of thekatgene-based tree was similar to those of trees obtained by analyzing the
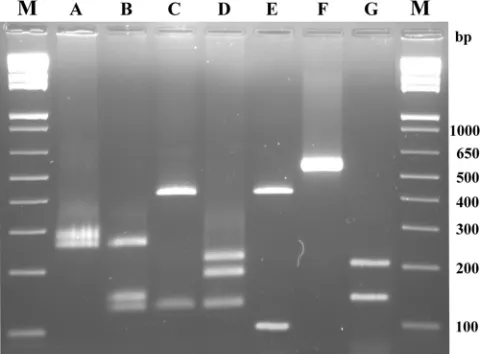
rpoB, sodA, tuf, gap, 16S rRNA, and hsp60 genes, recently published by Ghebremedhin et al. (16). Sequences from strains FIG. 2. RFLP analysis of 1,114-bp KDN-For/KDN-Rev PCR prod-ucts. Lanes: A, undigested product fromS. equorumsubsp. linensDSM 15097T; B, product fromS. equorum subsp. linens DSM 15097T di-gested by CfoI restriction enzyme; C, product fromS. xylosusDSM 20266 digested by CfoI restriction enzyme; D, undigested product from S. xylosusDSM 20266; E, product fromS. xylosusDSM 20266 digested by TaqI restriction enzyme; F, product fromS. saprophyticussubsp. saprophyticusDSM 20229T digested by CfoI restriction enzyme; G, product fromS. saprophyticus subsp. saprophyticus DSM 20229T di-gested by EcoRI restriction enzyme; H, undidi-gested product fromS. saprophyticussubsp. saprophyticus DSM 20229T; and M, 1 Kb Plus DNA ladder (Invitrogen).
on May 16, 2020 by guest
http://jcm.asm.org/
of the same species clustered very close (bootstrap values, 99 to 100%), with the exception of theS. saprophyticusDSM 20229
katB gene, clustering close to the katA genes of S. xylosus
strains (Fig. 1), due mainly to the presence of two catalase genes in these species. We named the previously knownkat
genekatAand the one discovered in this studykatB. Based on the analyses ofkatAfrom S. saprophyticusand katBfrom S. xylosus, these species are not very close.
Finally, different clusters with significant bootstrap values (⬎90%) could be identified (Fig. 1): (i) the S. saprophyticus
group, includingS. saprophyticus, S. xylosus, and S. equorum
(bootstrap value, 99%, based on thekatAandkatBgenes); (ii) theS. carnosusgroup, includingS. carnosus,S. condimenti, and
S. simulans (bootstrap value, 98%); (iii) the S. intermedius
group, including S. intermedius, S. pseudintermedius, and S. delphini(bootstrap value, 99%); (iv) theS. epidermidis/S. au-reus group, including S. epidermidis, S. capitis, S. caprae, S. haemolyticus, S. pasteuri, S. warneri, and S. aureus(bootstrap value, 93%); and (v) theS. sciurigroup, includingS. sciuri,S. vitulinus, andS. lentus(bootstrap value, 100%).
katgene sequence restriction polymorphism.In silico restric-tion (computer-aided) endonuclease analysis of all Staphylo-coccus katpartial sequences was performed by MacDNasis Pro (version 3.0.7.) software.
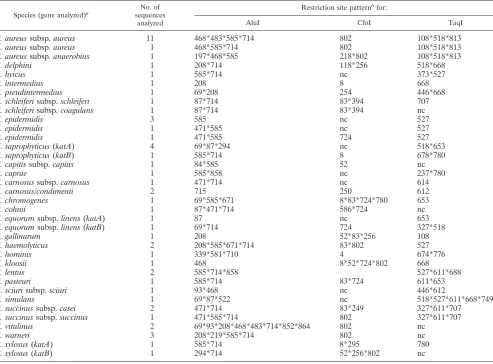
TaqI PCR-RFLPin silicopattern analysis may allow identi-fication and differentiation of the CPS species S. aureus, S. delphini,S. intermedius,S. pseudintermedius,S. schleiferisubsp.
coagulans, andS. hyicus(Table 2).
By coupling Alu with CfoI or TaqI PCR-RFLP in silico
patterns, all staphylococcal species could be differentiated (Ta-ble 2). In addition, CfoI and TaqI also allowed intraspecific polymorphism to be revealed. In particular, CfoI PCR-RFLP
in silico analysis provided discrimination between S. aureus
susbp.aureus and S. aureus susbp.anaerobius, as well as S. succinussubsp. caseiandS. succinussubsp. succinus, whereas by using TaqI, S. schleiferi subsp. schleiferi and S. schleiferi
subsp.coagulanscould be distinguished (Table 2).
[image:6.585.47.540.90.454.2]Identification and differentiation of CPS by the PCR-RFLP approach.To set up a rapid method allowing identification and differentiation of CPS strains, we designed universal primers TABLE 2. Positions of restriction sites of the AluI, CfoI, and TaqI endonucleases on the sequenced 900-bpkatfragments
from staphylococci analyzed in this study
Species (gene analyzed)a No. of
sequences analyzed
Restriction site patternbfor:
AluI CfoI TaqI
S. aureussubsp.aureus 11 468*483*585*714 802 108*518*813 S. aureussubsp.aureus 1 468*585*714 802 108*518*813 S. aureussubsp. anaerobius 1 197*468*585 218*802 108*518*813
S. delphini 1 208*714 118*256 518*668
S. hyicus 1 585*714 nc 373*527
S. intermedius 1 208 8 668
S. pseudintermedius 1 69*208 254 446*668
S. schleiferisubsp.schleiferi 1 87*714 83*394 707 S. schleiferisubsp.coagulans 1 87*714 83*394 nc
S. epidermidis 3 585 nc 527
S. epidermidis 1 471*585 nc 527
S. epidermidis 1 471*585 724 527
S. saprophyticus(katA) 4 69*87*294 nc 518*653
S. saprophyticus(katB) 1 585*714 8 678*780
S. capitissubsp.capitis 1 84*585 52 nc
S. caprae 1 585*858 nc 237*780
S. carnosussubsp.carnosus 1 471*714 nc 614
S. carnosus/condimenti 2 715 250 612
S. chromogenes 1 69*585*671 8*83*724*780 653
S. cohnii 1 87*471*714 586*724 nc
S. equorumsubsp.linens(katA) 1 87 nc 653
S. equorumsubsp.linens(katB) 1 69*714 724 327*518
S. gallinarum 1 208 52*83*256 108
S. haemolyticus 2 208*585*671*714 83*802 527
S. hominis 1 339*581*710 4 674*776
S. kloosii 1 468 8*52*724*802 668
S. lentus 2 585*714*858 527*611*688
S. pasteuri 1 585*714 83*724 611*653
S. sciurisubsp.sciuri 1 93*468 nc 446*612
S. simulans 1 69*87*522 nc 518*527*611*668*749
S. succinussubsp.casei 2 471*714 83*249 327*611*707 S. succinussubsp.succinus 1 471*585*714 802 327*611*707 S. vitulinus 2 69*93*208*468*483*714*852*864 802 nc
S. warneri 3 208*219*585*714 802 nc
S. xylosus(katA) 1 585*714 8*295 780
S. xylosus(katB) 1 294*714 52*256*802 nc
a
For species harboring two catalase genes, the sequenced gene is indicated.
b
Numbers are nucleotide positions. nc, no cut.
on May 16, 2020 by guest
http://jcm.asm.org/
CPSK1F and CPSK6R, allowing the amplification of a 544-bp region ofkatAcontaining polymorphic TaqI restriction sites of the various CPS strains. As shown in Fig. 3, clear differentia-tion of all species was achieved by using the PCR-RFLP ap-proach. However, to further validate and confirm the useful-ness of this assay, the PCR-RFLP patterns displayed by strains
listed in Table 1 were also determined and analyzed. Based on their PCR-RFLP patterns, strains were identified as follows. Among 56 strains received asS. aureus, 53 were confirmed to be S. aureus, 2 (SAT-51 and SAT-52) were identified as S. delphini, and 1 (SAT-48) was identified as S. intermedius. Among five strains received asS. delphini, three were confirmed to beS. delphiniand two (h-2c and P-27B) were identified asS. intermedius. Among 21 strains received asS. intermedius, 10 were confirmed to beS. intermedius, 1 strain (SAT-42) displayed a mixedS. aureus/S. intermedius pattern, 7 were identified asS. delphini, and 3 were identified asS. pseudintermedius. Identifica-tion of strains received asS. pseudintermedius,S. schleiferi, andS. hyicuswas confirmed. One strain received asS. chromogenes (Ter-24N) was identified asS. delphini. Finally, some strains received as
Staphylococcusspp. and showing patterns (designated X, Y, and Z in Table 1) different from those of CPS reference strains based on sequencing of thekatA gene (KDN-For/KDN-Rev PCR fragment) were identified asS. epidermidis(strains ED-1 and ED-2),S. chromogenes(strain SH4/03), and Staphylococ-cussp. (strain SH3/03, showing the highest level of similarity, 88%, toS. hyicus). To confirm the identification, some strains reclassified on the basis of their TaqI PCR-RFLP patterns were subjected to sequencing of thekatAgene (544-bp frag-ment). As indicated in Fig. 4, comparison of the resulting sequences with those from reference strains yielded findings in agreement with those of the PCR-RFLP analysis.
Moreover, the same strains were also subjected to partial sequencing of thegapgene (700-bp fragment) as described by Ghebremedhin et al. (16). As indicated in Fig. 5, identifications obtained by comparison of the resulting gap sequences with FIG. 3. TaqI RFLP analysis of 544-bp CPSK1F/CPSK6R PCR
[image:7.585.43.284.70.247.2]products. Patterns: A, product fromS. aureusDSM 20231T; B, product from S. delphini LMG 22190T; C, product from S. intermedius DSM20373T; D, product fromS. pseudintermediusLMG 22219T; E, product fromS. schleiferisubsp.schleiferiDSM 4807T; F, product from S. schleiferi subsp.coagulans DSM 6628T; and G, product fromS. hyicusDSM 20249T. M, 1 Kb Plus DNA ladder (Invitrogen).
FIG. 4. Neighbor-joining tree showing the phylogenetic relationships among some CPS strains andS. chromogenesandS. epidermidisbased on a comparison of 544-bpkatgene sequences. Bootstrap values based on 1,000 replications are given at the branching points when they are above 50%. The scale bar evaluates the sequence divergence.
on May 16, 2020 by guest
http://jcm.asm.org/
those from reference strains found in GenBank fit perfectly with the identifications obtained by PCR-RFLP analysis and
katAgene sequencing. The K1224 strain (of equine origin), originally identified asS. intermediusby traditional identifica-tion methods, was recently reclassified asS. delphinibased on
rpoBsequencing (showing 99.38% identity toS. delphini rpoB) (K. Becker, Mu¨nster University, Germany, personal commu-nication).
DISCUSSION
Due to the very high interspecies sequence similarity (90 to 99%) displayed by staphylococcal species, the use of 16S rRNA gene sequence analysis has been questioned in studies at the species level. Better results have been obtained by comparing sequences of other housekeeping genes such ashsp60, sodA,
rpoB,tuf, andgap(16).
In this study, we evaluated thekatgene as a new target for phylogenetic analysis of staphylococci and identification and differentiation of staphylococcal isolates at the species level.
Degenerate primers designed during this study were able to coamplify the two catalase genes of S. xylosus DSM 20266. Furthermore, by inverse PCR experiments, we were able to
determine the full nucleotide sequence ofkatBfrom this mi-croorganism. Two catalase genes were also found inS. equo-rumandS. saprophyticus. S. xylosus,S. equorum, andS. sapro-phyticus are the main staphylococcal species occurring in naturally fermented meat products (5–8).S. xylosusis widely used as a starter culture in association with lactic acid bacteria for process improvement (19). The presence of two catalase genes in the above-listed staphylococcal species may be a de-fense mechanism against hydrogen peroxide produced by lactic acid bacteria during fermentation.
Thekatgene sequences were less conserved (with interspe-cies sequence similarity ranging from 40 to 93%) than those of the genes rpoB (71.6 to 93.6% similar), hsp60 (74 to 93% similar), andtuf (86 to 97% similar) and slightly more con-served than those of thegapgene (24 to 96% similar) (16).
Thekat-based tree indicates the divergence of staphylococ-cal species and was well supported for all the strains analyzed during this study. In addition, with bothkatgenes and a boot-strap value of⬎90%, the staphylococcal species were divided into five well-supported clusters: theS. saprophyticus group, theS. carnosus/S. simulansgroup, theS. intermediusgroup, the
[image:8.585.109.472.74.419.2]S. epidermidis/S. aureusgroup, and theS. sciurigroup. Similar but not identical results were obtained by Ghebremedhin et al. FIG. 5. Neighbor-joining tree showing the phylogenetic relationships among some CPS strains andS. chromogenesandS. epidermidisbased on a comparison of 700-bpgapgene sequences. Bootstrap values based on 1,000 replications are given at the branching points when they are above 50%. The scale bar evaluates the sequence divergence.
on May 16, 2020 by guest
http://jcm.asm.org/
(16), who analyzedgapgenes and divided staphylococcal spe-cies into only four well-supported clusters: theS. sciurigroup, theS. hyicus/S. intermediusgroup, theS. haemolyticus/S. simu-lansgroup, and theS. epidermidis/S. aureusgroup. Therefore,
katgene analysis may represent an additional marker for in-ferring staphylococci phylogenetics.
katAgenes display a high level of restriction endonuclease polymorphism, offering good opportunities for rapid, accurate species-level identification of staphylococcal isolates. However, as indicated by ourin silicoanalysis ofkatAgenes from only reference strains of 26 staphylococcal species, sequential use of two endonucleases, first AluI and then CfoI or TaqI, is needed to achieve identification of staphylococcal isolates at the spe-cies and subspespe-cies levels. In contrast, by selecting the suitable
katAregion to analyze, we demonstrated with our evaluation of about 100 wild strains that TaqI PCR-RFLP alone can discriminate among CPS species.
Several studies (11, 29) have highlighted biases in phenotyp-ically differentiating the various CPS species. Sasaki et al. (29), for example, reclassified some phenotypically identifiedS. in-termediusisolates asS. pseudintermediusorS. delphiniby se-quence analyses ofsodA, hsp60, and nucgenes. Commercial kits for identifyingS. delphiniandS. pseudintermedius, the most recently described CPS species, are not available. Therefore, at present the most appropriate approach to reliably identify the latter two species entails sequencing of more than one house-keeping gene:gap,sodA,hsp60, and/ornuc.
In this study, we designed a robust, rapid, and low-cost approach (requiring about 6 h from DNA isolation to the production of results and⬍5 Euros per strain tested) to iden-tify and differentiate CPS species, based on TaqI restriction endonuclease analysis of the 544-bp PCR-amplifiedkatAgene fragment (TaqI PCR-RFLP analysis).
Our strategy is similar to other PCR-RFLP methods based ongap,dnaJ, andptagenes (2, 20, 38, 39). However, Yugueros et al. (38, 39) analyzed only three CPS species (S. aureus, S. intermedius, and S. delphini), Hauschild and Stepanovic (20) obtained good results by using two restriction enzymes sequen-tially but did not analyzeS. delphinistrains, and Bannoehr et al. (2) were unable to differentiate strains of S. delphini, S. intermedius, andS. schleiferi.
Our PCR-RFLP technique was able to differentiate all CPS species. This approach was validated by unambiguous identi-fication of more than 100 strains, including reference and wild CPS strains isolated from different sources. Owing to its spec-ificity, manageability, and rapidity, thekat-based PCR-RFLP approach proposed in this study can be considered a valid strategy for rapid identification of CPS strains at the species level.
In conclusion, the high variability of thekatnucleotide se-quences allows discrimination among closely related species, opens new possibilities for rapid, reliable identification of staphylococci, and offers good opportunities to develop assays based on hybridization (probe design), PCR (primer design), or DNA chip (microarray design) technologies. Upon compar-ing our results with the recent findcompar-ings of Ghebremedhin et al. (16), the greater usefulness ofkat gene analysis than of 16S rRNA,rpoB,hsp60, andtufgene analyses is evident. Moreover, for several species of bacteria, nucleotide sequence analysis of multiple-protein-encoding loci has led to reliable phylogenies
that have improved our understanding of the population struc-ture. A multigenic approach fulfils the recent recommenda-tions of the ad hoc committee for the reevaluation of the definition of bacterial species (32). Thekatgene may be con-sidered an excellent molecular marker for inferring the taxon-omy and phylogeny of members of the genusStaphylococcus
and may represent a good candidate in multilocus schemes designed to identify and characterize staphylococci.
ACKNOWLEDGMENTS
We thank G. Lina of the CNTS (Lyon, France), K. Becker of the IMM (Mu¨nster, Germany), K. Hiramatsu of the DICS (Tokyo, Japan), and A. Ianieri of the DSA (Teramo, Italy) for kindly providing strains used in this study. Thanks are also due to Giancarlo Ciao for his technical assistance.
This work was supported partly by an Italian MIUR grant.
REFERENCES
1.Altschul, S. F., T. L. Madden, A. A. Schaffer, J. Zhang, Z. Zhang, W. Miller, and D. J. Lipman.1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res.25:3389–3402. 2.Bannoehr, J., A. Franco, M. Iurescia, A. Battisti, and R. Fitzgerald.2009.
Molecular diagnostic identification of Staphylococcus pseudintermedius. J. Clin. Microbiol.47:469–471.
3.Barrie`re, C., R. Bru¨ckner, D. Centano, and R. Talon.2002. Characterization of thekatA gene encoding a catalase and evidence for at least a second catalase activity inStaphylococcus xylosus, bacteria used in food fermenta-tion. FEMS Microbiol. Lett.216:277–283.
4.Becker, K., G. Haverkamper, C. von Eiff, R. Roth, and G. Peters.2001. Survey of staphylococcal enterotoxin genes, exfoliative toxin genes, and toxic shock syndrome toxin 1 gene in non-Staphylococcus aureusspecies. Eur. J. Clin. Microbiol. Infect. Dis.20:407–409.
5.Blaiotta, G.2002. Polyphasic molecular approach to identification of staph-ylococci in food ecosystems. PhD thesis. Department of Food Science, Uni-versity of Naples Federico II, Naples, Italy.
6.Blaiotta, G., C. Pennacchia, D. Ercolini, G. Moschetti, and F. Villani.2003. Combining denaturing gradient gel electrophoresis of 16S rDNA V3 region and 16S–23S rDNA spacer region polymorphism analyses for the identifica-tion of staphylococci from Italian fermented sausages. Syst. Appl. Microbiol.
26:423–433.
7.Blaiotta, G., C. Pennacchia, E. Parente, and F. Villani.2003. Design and evaluation of specific PCR primers for rapid and reliable identification of
Staphylococcus xylosusstrains isolated from dry fermented sausages. Syst. Appl. Microbiol.26:601–609.
8.Blaiotta, G., C. Pennacchia, F. Villani, A. Ricciardi, R. Tofalo, and E. Parente.2004. Biological diversity of staphylococci isolated from traditional fermented sausages produced in Basilicata (Southern Italy). J. Appl. Micro-biol.97:271–284.
9.Blaiotta, G., D. Ercolini, C. Pennacchia, V. Fusco, A. Casaburi, O. Pepe, and F. Villani.2004. PCR detection of staphylococcal enterotoxin genes in Staph-ylococcusspp. strains isolated from meat and dairy products. Evidence for new variants of seG andseI in S. aureus AB-8802. J. Appl. Microbiol.
97:719–730.
10.Blaiotta, G., V. Fusco, C. von Eiff, F. Villani, and K. Becker.2006. Biotyping of enterotoxigenicStaphylococcus aureusby enterotoxin gene cluster (egc) polymorphism andspatyping analyses. Appl. Environ. Microbiol.72:6117– 6123.
11.Delmas, J., J. P. Chacormarc, F. Robin, P. Giammarinaro, R. Talon, and R. Bonnet.2008. Evaluation of the Vitek 2 system with a variety of staphylo-coccal species. J. Clin. Microbiol.46:311–313.
12.Devriese, L. A., K. Hermans, M. Baele, and F. Haesebrouck.2008. Staphy-lococcus pseudintermediusversusStaphylococcus intermedius. Vet. Microbiol.
133:206–207.
13.Drancourt, M., and D. Raoult.2002.rpoBgene sequence-based identifica-tion ofStaphylococcusspecies. J. Clin. Microbiol.40:1333–1338. 14.Edwards, K., M. E. Kaufmenn, and N. A. Saunders.2001. Rapid and
accu-rate identification of coagulase-negative staphylococci by real-time PCR. J. Clin. Microbiol.39:3047–3051.
15.Euzeby, J. P.1997. List of bacterial names with standing in nomenclature: a folder available on the internet. Int. J. Syst. Bacteriol.47:590–592. 16.Ghebremedhin, B., F. Layer, W. Konig, and B. Konig.2008. Genetic
classi-fication and distinguishing ofStaphylococcusspecies based on different par-tialgap, 16S rRNA,hsp60,rpoB,sodA, andtufgene sequences. J. Clin. Microbiol.46:1019–1025.
17.Goh, S. H., Z. Santucci, W. E. Kloos, M. Faltyn, C. G. George, D. Driedger, and S. M. Hemmingsen.1997. Identification ofStaphylococcusspecies and
on May 16, 2020 by guest
http://jcm.asm.org/
subspecies by the chaperonin 60 gene identification method and reverse checkerboard hybridization. J. Clin. Microbiol.35:3116–3121.
18.Gory, L., L. Millet, J. J. Godon, and M. C. Montel.1999. Identification of
Staphylococcus carnosusandStaphylococcus warneriisolated from meat by florescentin situhybridization with 16S-targeted oligonucleotide probes. Syst. Appl. Microbiol.22:225–228.
19.Hammes, W. P., and C. Hertel.1998. New developments in meat starter cultures. Meat Sci.49:125–138.
20.Hauschild, T., and S. Stepanovic.2008. Identification ofStaphylococcusspp. by PCR-restriction fragment length polymorphism analysis ofdnaJgene. J. Clin. Microbiol.46:3875–3879.
21.Kloos, W. E.1990. Systematics and the natural history of staphylococci 1. Soc. Appl. Bacteriol. Symp. Ser.19:25S–37S.
22.Martineau, F., F. J. Picard, D. Ke, S. Paradis, P. H. Roy, M. Ouellette, and M. G. Bergeron.2001. Development of a PCR assay for identification of staphylococci at genus and species levels. J. Clin. Microbiol.39:2541–2547. 23.Mauriello, G., A. Casaburi, G. Blaiotta, and F. Villani.2004. Isolation and technological properties of coagulase negative staphylococci from fermented sausages of southern Italy. Meat Sci.67:149–158.
24.Nychas, G. J. E., and J. S. Arkoudelos.1990. Staphylococci: their role in fermented sausages. Soc. Appl. Bacteriol. Symp. Ser.69:167S–188S. 25.Park, B., V. Nizet, and G. Y. Liu. 2008. Role ofStaphylococcus aureus
catalase in niche competition againstStreptococcus pneumoniae. J. Bacteriol.
190:2275–2278.
26.Poyart, C., G. Quesne, C. Boumaila, and P. Trieu-Cout.2001. Rapid and accurate specific-level identification of coagulase-negative staphylococci by usingsodAgene as a target. J. Clin. Microbiol.39:4296–4331.
27.Saitou, N., and M. Nei.1987. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol.4:406–425. 28.Sanz, R., I. Marin, J. A. Ruiz-Santa-Quiteria, J. A. Orden, D. Cid, R. D. Diez,
K. S. Silhadi, R. Amil, and R. de la Fuente.2000. Catalase deficiency in
Staphylococcus aureussubsp.anaerobiusis associated with natural loss-of-function mutations within the structural gene. Microbiology146:465–475. 29.Sasaki, T., K. Kikuchi, Y. Tanaka, N. Takahashi, S. Kamata, and K.
Hira-matsu.2007. Reclassification of phenotypically identifiedStaphylococcus in-termediusstrains. J. Clin. Microbiol.45:2770–2778.
30.Shah, M. M., H. Iihara, M. Noda, S. X. Song, P. H. Nhung, K. Ohkusu, Y.
Kawamura, and T. Ezaki.2007.dnaJgene sequence-based assay for species identification and phylogenetic grouping in the genusStaphylococcus. Int. J. Syst. Evol. Microbiol.57:25–30.
31.Soriano, J. M., G. Font, H. Rico, J. C. Molto, and J. Manes.2002. Incidence of enterotoxigenic staphylococci and their toxins in foods. J. Food Prot.
65:857–860.
32.Stackebrandt, E., W. Frederiksen, G. M. Garrity, et al.2002. Report of the ad hoc committee for the re-evaluation of the species definition in bacteri-ology. Int. J. Syst. Evol. Microbiol.52:1043–1047.
33.Tamura, K., J. Dudley, M. Nei, and S. Kumar.2007. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol. Biol. Evol.24:1596–1599.
34.Thompson, J. D., D. G. Higgins, and T. J. Gibson.1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res.22:4673–4680.
35.Van Hoovels, L., A. Vankeerberghen, A. Boel, K. Van Vaerenbergh, and H. De Beenhouwer.2006. First case ofStaphylococcus pseudintermedius infec-tion in a human. J. Clin. Microbiol.44:4609–4612.
36.Vannuffel, P., M. Heusterspreute, M. Bouyer, B. Vandercam, M. Philippe, and J. L. Gala.1999. Molecular characterization offemA from Staphylococ-cus hominisandStaphylococcus saprophyticus, andfemA-based discrimina-tion of staphylococcal species. Res. Microbiol.150:129–141.
37.Von Eiff, C., C. R. Arciola, L. Montanaro, K. Becker, and D. Campoccia.
2006. EmergingStaphylococcusspecies as new pathogens in implant infec-tions. Int. J. Artific. Organs29:360–367.
38.Yugueros, J., A. Temprano, B. Berzal, M. Sanchez, C. Hernanz, J. M. Luengo, and G. Naharro. 2000. Glyceraldehyde-3-phosphate dehydroge-nase-encoding gene as a useful taxonomic tool for Staphylococcus spp. J. Clin. Microbiol.38:4351–4355.
39.Yugueros, J., A. Temprano, M. Sanchez, C. Hernanz, J. M. Luengo, and G. Naharro. 2001. Identification of Staphylococcus spp. by PCR restriction fragment length polymorphism ofgapgene. J. Clin. Microbiol.39:3693–3695. 40.Zell, C., M. Resch, R. Rosenstein, T. Albrecht, C. Hertel, and F. Gotz.2008. Characterization of toxin production of coagulase-negative staphylococci isolated from foods and starter cultures. Int. J. Food Microbiol.127:246–251.