N
-(Ferrocenylmethyl)dodecan-1-amine
Li-Zhen Sun, Wei An, Hua-Cheng Zhang, Fei-Fei Xin and Ai-You Hao*
School of Chemistry and Chemical Engineering, Shandong University, Jinan 250100, People’s Republic of China
Correspondence e-mail: [email protected] Received 23 January 2010; accepted 9 February 2010
Key indicators: single-crystal X-ray study;T= 293 K; mean(C–C) = 0.003 A˚;
Rfactor = 0.037;wRfactor = 0.112; data-to-parameter ratio = 21.5.
The title compound, [Fe(C5H5)(C18H32N)], was synthesized by
the amination of ferrocenecarbaldehyde. In the complex, the two cyclopentadienyl (Cp) rings are almost parallel with a dihedral angle of 1.36 (8), and are separated by a centroid–
centroid distance of 3.299 (2) A˚ . In the crystal, adjacent molecules are linked into a one-dimensional supramolecular structureviaweak C—H interactions between the Cp ring H atom and the Cp ring.
Related literature
For the applications of ferrocene in drug design, see: Attekeet al. (2003); Baramee et al. (2006). For linear ferrocene compounds in supramolecular chemisty, see: Zhang et al.
(2010). For a related structure, see: Zheng & Liu (2009).
Experimental
Crystal data
[Fe(C5H5)(C18H32N)]
Mr= 383.39 Monoclinic,P21=c a= 26.7005 (6) A˚ b= 8.0549 (2) A˚ c= 10.0069 (2) A˚
= 97.534 (1)
V= 2133.60 (8) A˚3
Z= 4
MoKradiation
= 0.71 mm1 T= 293 K
0.300.260.18 mm
Data collection
Bruker APEXII CCD diffractometer
Absorption correction: multi-scan (SADABS; Sheldrick, 1996) Tmin= 0.815,Tmax= 0.881
20623 measured reflections 4900 independent reflections 3892 reflections withI> 2(I) Rint= 0.021
Refinement
R[F2> 2(F2)] = 0.037 wR(F2) = 0.112
S= 1.09 4900 reflections
228 parameters
H-atom parameters constrained max= 0.44 e A˚
3
min=0.37 e A˚
[image:1.610.316.567.168.214.2]3
Table 1
Selected bond lengths (A˚ ).
C14—Fe1 2.0502 (16)
C15—Fe1 2.0501 (18)
C16—Fe1 2.038 (2)
C17—Fe1 2.0295 (18)
C18—Fe1 2.0398 (18)
C19—Fe1 2.038 (2)
C20—Fe1 2.0350 (17)
C21—Fe1 2.0375 (18)
C22—Fe1 2.0398 (18)
[image:1.610.313.564.290.326.2]C23—Fe1 2.0347 (19)
Table 2
Hydrogen-bond geometry (A˚ ,). Cg1 is the centroid of the C19–C23 ring.
D—H A D—H H A D A D—H A
C18—H18 Cg1i
0.98 2.96 3.870 (2) 155
Symmetry code: (i)x;yþ5 2;zþ
1 2.
Data collection:APEX2(Bruker, 2007); cell refinement: SAINT-Plus(Bruker, 2007); data reduction:SAINT-Plus; program(s) used to solve structure:SIR97 (Altomareet al., 1999); program(s) used to refine structure:SHELXL97(Sheldrick, 2008); molecular graphics: SHELXTL(Sheldrick, 2008) andDIAMOND(Brandenburg, 1999); software used to prepare material for publication:WinGX(Farrugia, 1999).
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: HY2280).
References
Altomare, A., Burla, M. C., Camalli, M., Cascarano, G. L., Giacovazzo, C., Guagliardi, A., Moliterni, A. G. G., Polidori, G. & Spagna, R. (1999).J. Appl. Cryst.32, 115–119.
Atteke, C., Me Ndong, J. M., Aubouy, A., Maciejewski, L., Brocard, J., Lebibi, J. & Deloron, P. (2003).J. Antimicrob. Chemother.51, 1021–1024. Baramee, A., Coppin, A., Mortuaire, M., Pelinski, L., Tomavoc, S. & Brocard,
J. (2006).Bioorg. Med. Chem.14, 1294–1302.
Brandenburg, K. (1999).DIAMOND.Crystal Impact GbR, Bonn, Germany. Bruker (2007). APEX2 and SAINT-Plus. Bruker AXS Inc., Madison,
Wisconsin, USA.
Farrugia, L. J. (1999).J. Appl. Cryst.32, 837–838.
Sheldrick, G. M. (1996).SADABS. University of Go¨ttingen, Germany. Sheldrick, G. M. (2008).Acta Cryst.A64, 112–122.
Zhang, H., An, W., Liu, Z., Hao, A., Hao, J., Shen, J., Zhao, X., Sun, H. & Sun, L. (2010).Carbohydr. Res.345, 87–96.
Zheng, X.-L. & Liu, J.-T. (2009).Acta Cryst.E65, m432. Acta Crystallographica Section E
Structure Reports
Online
supporting information
Acta Cryst. (2010). E66, m301 [doi:10.1107/S1600536810005155]
N
-(Ferrocenylmethyl)dodecan-1-amine
Li-Zhen Sun, Wei An, Hua-Cheng Zhang, Fei-Fei Xin and Ai-You Hao
S1. Comment
The high lipophilicity and electrochemical behaviour of ferrocene render it very attractive for drug design. The
incorporation of a ferrocenyl moiety into the 'standard′ drug offers new possibilities in therapeutic applications and
reversal of drug resistances (Atteke et al., 2003; Baramee et al., 2006). On the other hand, the linear compounds derived
from ferrocene have shown great useful functions in supramolecular chemisty (Zhang et al., 2010). In this paper, we
report the synthesis and crystal structure of the title compound.
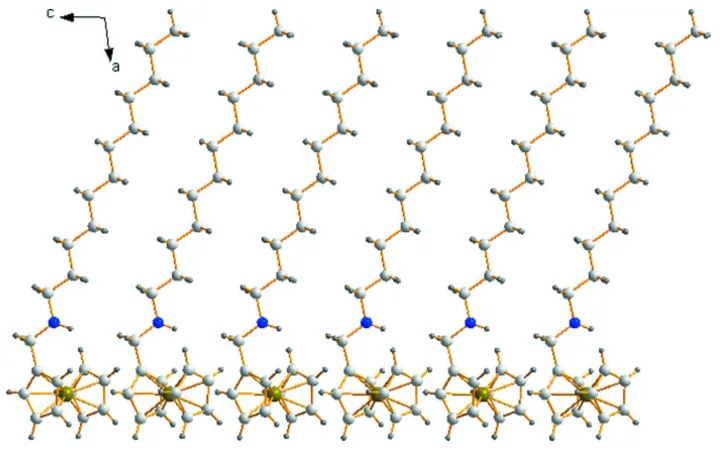
As shown in Fig. 1, the title compound has a tadpole-typed molecular configuration. The planes of two
cyclo-pentadienyl (Cp) rings in the ferrocene scaffold are approximately parallel with a centroid–centroid distance of 3.299 (2)
Å and a dihedral angle of 1.36 (8)°. The Fe—C bond distances are in the range of 2.0295 (18) to 2.0502 (16) Å (Table 1),
which are consistent with those data in the similar compounds previously reported (Zheng & Liu, 2009). The adjacent
molecules are linked by weak C—H···π interactions between ferrocene cores (Table 2), forming a one-dimensional
supramolecular architecture as shown in Fig. 2.
S2. Experimental
A mixture of ferrocenecarbaldehyde (2.415 g, 0.01 mol) and lauryl amine (0.08 mol) in anhydrous methanol (50 ml) was
stirred and kept refluxing for 12 h under a nitrogen atmosphere in dark place. NaBH4 (0.110 g, 2.9 mmol) was added in
small portions. The mixture was stirred for 15 h, and 10 ml acetone was added to stop the reaction. The mixture was
extracted with CH2Cl2 (30 ml) 10 min later. The combined organic phase was dried with anhydrous Na2SO4, and then
evaporated in vacuo. The residue was purified by silica gel column chromatography (eluent, petroleum ether : ethyl
acetate = 5:1 by volume) to give the title compound as yellow powder (yield 1.545 g, 80.7%). Single crystals were grown
in a mixed solution of hexane/ethyl acetate (2:1 by volume) at room temperature.
S3. Refinement
H atoms were positioned geometrically and refined as riding atoms, with C—H = 0.98 (CH), 0.97 (CH2) and 0.96 (CH3)
Figure 1
[image:3.610.126.486.104.332.2]Molecular structure of the title compound. Displacement ellipsoids are drawn at the 50% probability level.
Figure 2
A packing diagram of the title compound along the b axis.
N-(Ferrocenylmethyl)dodecan-1-amine
Crystal data
[Fe(C5H5)(C18H32N)] Mr = 383.39
Monoclinic, P21/c
Hall symbol: -P 2ybc a = 26.7005 (6) Å b = 8.0549 (2) Å c = 10.0069 (2) Å β = 97.534 (1)° V = 2133.60 (8) Å3 Z = 4
F(000) = 832 Dx = 1.194 Mg m−3
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 8358 reflections θ = 2.3–27.4°
µ = 0.71 mm−1 T = 293 K Prism, orange
0.30 × 0.26 × 0.18 mm
Data collection
Bruker APEXII CCD diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
φ and ω scans
Absorption correction: multi-scan (SADABS; Sheldrick, 1996) Tmin = 0.815, Tmax = 0.881
20623 measured reflections 4900 independent reflections 3892 reflections with I > 2σ(I) Rint = 0.021
θmax = 27.5°, θmin = 0.8° h = −34→34
Refinement
Refinement on F2
Least-squares matrix: full R[F2 > 2σ(F2)] = 0.037 wR(F2) = 0.112 S = 1.09 4900 reflections 228 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H-atom parameters constrained w = 1/[σ2(F
o2) + (0.0649P)2 + 0.2305P]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max = 0.002
Δρmax = 0.44 e Å−3
Δρmin = −0.37 e Å−3
Extinction correction: SHELXL, Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4
Extinction coefficient: 0.0051 (7)
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
H11A 0.1930 0.7717 0.9335 0.067* H11B 0.1953 0.9653 0.9216 0.067* C12 0.21815 (6) 0.8834 (3) 1.11265 (18) 0.0558 (4) H12A 0.2132 0.9901 1.1540 0.067* H12B 0.2076 0.7976 1.1709 0.067* C13 0.30453 (6) 0.8840 (3) 1.22683 (17) 0.0544 (4) H13A 0.2953 0.8050 1.2925 0.065* H13B 0.2998 0.9948 1.2611 0.065* C14 0.35911 (6) 0.8607 (2) 1.21065 (17) 0.0483 (4) C15 0.37909 (7) 0.7502 (2) 1.12038 (19) 0.0560 (4) H15 0.3594 0.6787 1.0537 0.067* C16 0.43262 (8) 0.7632 (3) 1.1426 (2) 0.0698 (6) H16 0.4563 0.7016 1.0943 0.084* C17 0.44560 (7) 0.8800 (3) 1.2456 (2) 0.0731 (6) H17 0.4800 0.9137 1.2817 0.088* C18 0.40041 (7) 0.9412 (3) 1.28863 (18) 0.0591 (5) H18 0.3981 1.0241 1.3593 0.071* C19 0.39885 (9) 1.2394 (3) 1.0622 (2) 0.0687 (6) H19 0.3971 1.3232 1.1325 0.082* C20 0.35736 (7) 1.1625 (2) 0.98373 (19) 0.0597 (5) H20 0.3216 1.1831 0.9899 0.072* C21 0.37662 (8) 1.0532 (3) 0.89473 (19) 0.0596 (5) H21 0.3566 0.9829 0.8279 0.071* C22 0.42930 (8) 1.0612 (3) 0.9174 (2) 0.0639 (5) H22 0.4525 0.9971 0.8693 0.077* C23 0.44322 (7) 1.1742 (3) 1.0199 (2) 0.0680 (6) H23 0.4779 1.2043 1.0563 0.082* Fe1 0.401688 (8) 0.98868 (3) 1.08888 (2) 0.04257 (11) N1 0.27153 (5) 0.8617 (2) 1.10053 (15) 0.0577 (4) H1 0.2825 0.8379 1.0258 0.069*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
C15 0.0598 (11) 0.0504 (10) 0.0601 (11) 0.0051 (8) 0.0169 (9) 0.0106 (8) C16 0.0587 (12) 0.0736 (13) 0.0800 (15) 0.0258 (10) 0.0200 (10) 0.0332 (12) C17 0.0439 (10) 0.1064 (18) 0.0655 (13) 0.0029 (11) −0.0057 (9) 0.0377 (13) C18 0.0569 (11) 0.0795 (12) 0.0391 (9) −0.0042 (10) −0.0007 (8) 0.0119 (9) C19 0.1042 (18) 0.0467 (10) 0.0548 (11) 0.0008 (10) 0.0089 (11) −0.0005 (8) C20 0.0475 (9) 0.0644 (11) 0.0671 (12) 0.0135 (9) 0.0077 (9) 0.0209 (10) C21 0.0716 (13) 0.0614 (11) 0.0421 (9) 0.0004 (10) −0.0060 (9) 0.0084 (9) C22 0.0663 (12) 0.0721 (12) 0.0580 (12) 0.0155 (11) 0.0253 (10) 0.0193 (10) C23 0.0512 (10) 0.0744 (13) 0.0760 (14) −0.0130 (10) −0.0002 (10) 0.0244 (11) Fe1 0.03650 (15) 0.05241 (17) 0.03810 (16) 0.00326 (9) 0.00232 (10) 0.00551 (9) N1 0.0407 (7) 0.0843 (11) 0.0486 (8) −0.0016 (7) 0.0081 (6) −0.0071 (8)
Geometric parameters (Å, º)
C12—N1 1.457 (2) N1—H1 0.8600 C12—H12A 0.9700
C22—C21—Fe1—C17 −41.8 (3) C18—C14—Fe1—C19 −77.42 (14) C20—C21—Fe1—C17 −161.1 (2) C13—C14—Fe1—C19 41.90 (19) C22—C21—Fe1—C23 37.06 (13) C15—C14—Fe1—C16 −37.61 (12) C20—C21—Fe1—C23 −82.23 (13) C18—C14—Fe1—C16 81.63 (14) C22—C21—Fe1—C20 119.30 (18) C13—C14—Fe1—C16 −159.05 (19) C22—C21—Fe1—C19 81.10 (14) C15—C14—Fe1—C18 −119.24 (16) C20—C21—Fe1—C19 −38.20 (12) C13—C14—Fe1—C18 119.3 (2) C22—C21—Fe1—C16 −76.83 (16) C15—C14—Fe1—C22 44.6 (2) C20—C21—Fe1—C16 163.88 (12) C18—C14—Fe1—C22 163.9 (2) C22—C21—Fe1—C18 162.9 (2) C13—C14—Fe1—C22 −76.8 (3) C20—C21—Fe1—C18 43.6 (3) C18—C14—Fe1—C15 119.24 (16) C20—C21—Fe1—C22 −119.30 (18) C13—C14—Fe1—C15 −121.4 (2) C22—C21—Fe1—C15 −119.41 (13) C14—C13—N1—C12 179.95 (15) C20—C21—Fe1—C15 121.30 (12) C11—C12—N1—C13 174.70 (16)
Hydrogen-bond geometry (Å, º)
Cg1 is the centroid of the C19–C23 ring.
D—H···A D—H H···A D···A D—H···A
C18—H18···Cg1i 0.98 2.96 3.870 (2) 155