Vanadium Hydride as Conversion Type Negative Electrode for All-Solid-State
Lithium-Ion-Battery
Yasuhiro Matsumura
1,+1, Keiji Takagishi
2, Hiroki Miyaoka
3and Takayuki Ichikawa
1,3,4,+21Graduate School of Integrated Arts and Sciences, Hiroshima University, Higashi-Hiroshima 739-8521, Japan
2Graduate School of Advanced Sciences of Matter, Hiroshima University, Higashi-Hiroshima 739-8530, Japan
3Natural Science Center for Basic Research and Development, Hiroshima University, Higashi-Hiroshima 739-8530, Japan
4Graduate School of Engineering, Hiroshima University, Higashi-Hiroshima 739-8527, Japan
Possibility as an electrode by using vanadium hydride, which is one of typical metal hydrides as hydrogen storage alloy, was examined for an all solid lithium ion battery. The results obtained show vanadium hydride as a negative electrode material by conversion reaction were clarified for thefirst time in this work. By analyzing the charging/discharging properties and XRD profiles for each process, the reaction mechanism of the conversion reaction of vanadium system and lithiation reaction of hard carbon was clarified.
[doi:10.2320/matertrans.MT-M2019105]
(Received April 3, 2019; Accepted June 28, 2019; Published August 9, 2019)
Keywords: lithium ion battery, hydride negative electrode material, vanadium hydride, conversion reaction, solid state electrolyte, lithium borohydride
1. Introduction
Electrode materials such as silicon (Si) and metal hydrides (MH) with higher capacity than graphite have been actively studied.110) In 2008, Oumellal et al. reported that magne-sium hydride (MgH2) can be used as a negative electrode material for LIBs using organic liquid electrolyte.11) The theoretical capacity of MgH2 is 2034 mAh/g, which is significantly higher than that of graphite intercalation mentioned above. The reaction of metal hydride with lithium ions proceeds through the following reaction and termed as conversion reaction,
MþLiH,MHxþxLiþþxe: ð1Þ
For the practical development of metal hydride electrode, the understanding of reaction mechanism is necessary in order to improve the properties such as cyclic performance, over-potential, and kinetics. In addition to the electrode materials, investigation on the electrolyte itself is also important to safely utilize LIBs. In the case of conventional LIBs using the organic liquid electrolyte, there is a risk offire accidents due to the leakage of the liquid electrolyte. In recent years, solid state inorganic electrolytes have attracted the attention of scientific community. The sulfur-based solid electrolyte shows excellent lithium ion conductivity at room temper-ature.1216) Kanno et al. have synthesized thio-LISICON which also shows the high ion conductivity at room temperature.1721) Besides, Matsuo et al. recently reported that the complex hydride LiBH4, which is well-known as a promising hydrogen storage material reveals super ionic conductor above a phase transition at 115°C.2228)Thus, all solid state lithium ion batteries using metal hydride electrodes and LiBH4 electrolyte are expected as a next generation electric storage device because of the high capacity and safety.29) Liang et al. reported the excellent
electrochemical performance and the mechanism of con-version reaction in all solid state LIB composed of MgH2as electrode and LiBH4as electrolyte.30,31)Moreover, Kawahito et al. investigated the electrochemical properties of TiH2 using the LiBH4 electrolyte.32) Although the metal hydride is recognized as a potential and interesting electrode material to establish high performance batteries as seen above, there are only few reports on the other hydrides. In this paper, vanadium hydride is focused as the promising negative electrode material for the all solid state lithium ion batteries. Vanadium (V), having a bcc structure, is one of the attractive hydrogen storage materials. It can absorb and release 3.8 mass%of hydrogen3342)via following absorption and desorption reactions,
2Vð¡Þ þ1=2H2,V2Hð¢1Þ ð2Þ V2Hð¢1Þ þ1=2H2,2VHð¢2Þ ð3Þ VHð¢2Þ þ1=2H2,VH2ð£Þ ð4Þ
where the ¢1 phase formed at first reaction step is thermodynamically stable even in the air atmosphere at room temperature. However, the £ phase as the final hydrogenated state is unstable phase and releases hydrogen easily without hydrogen partial pressure at room temper-ature.33,43,44)
On the other hand, the conversion reaction of vanadium (V) and lithium hydride (LiH) can be assumed as follows:
VþxLiH,VHxþxLi ð5Þ
From the Nernst equation, theoretical reaction potential corresponding to each hydride phase can be obtained as follows.
E¼ 1
xFðHðVHxÞ TðSðVHxÞ SðVÞÞ
þxTðSðLiHÞ SðLiÞÞ xHðLiHÞÞ ð6Þ where the enthalpy and entropy values corresponding to LiH are obtained from a databook and the values corresponding to VHxshould be obtained from PC isothermal profiles of +1Graduate Student, Hiroshima University
+2Corresponding author, E-mail: tichi@hiroshima-u.ac.jp
VHx. According to thermodynamic considerations, the total
conversion reaction between V and LiH can be written as follows:
Vþ2LiH$VH2þ2Liþþ2e ð7Þ
where the theoretical reaction voltage and the capacity are obtained as 0.72 V and 1013 mAh/g. In this work, the electrochemical properties of V+LiH as the electrode material for all solid state LIB were investigated. A possible reaction mechanism is proposed.
2. Experiments
Since VH2 is an unstable material under an ambient conditions, a mixture of V and LiH was used as starting materials. First of all, V powder (99.9%, Rare metallic) and LiH powder (99.4%, Alfa Aesar) were mixed by mechanical ball-milling method with the following procedure. V and LiH were weighted in a molar ratio of 1:3 to be 300 mg in total. This mixture was mechanically milled for 2 h with 370 rpm by a planetary ball-milling apparatus (Fritsch, P7). The purchased LiBH4(½95%in purity, Sigma Aldrich) was hand milled in a mortar for 15 min in order to increase the interface with the electrode material. Then, LiBH4and acetylene black were heated at 200°C for 12 h under a dynamic vacuum in order to remove moisture adsorbed on the surface before further mixing. Finally, the V-LiH mixture was mixed with LiBH4as a Li-ion conducting material and acetylene black as an electron conducting material in a weight ratio of 40:30:30 by the ball-milling for 2 h with 370 rpm under 0.1 MPa of Ar. Li foil as a counter electrode and LiBH4 as a solid electrolyte were placed on a circular stainless-steel plate and transferred into a pellet maker and pressed at 10 MPa for 5 min. Then, the synthesized V-LiH electrode sample (about 10 mg) was spread on LiBH4 and pressed at 40 MPa for 5 min. The obtained pellet was sealed in coin type cell.
Charge and discharge properties were investigated by electrochemical test apparatus (HJ1001SD8, Hokuto Denko
Co.). For the electrochemical measurements, voltage window was fixed to 0.0051.0 V, and current density was kept as 10 mA/g (0.05 C). The battery tests were carried out at 125°C in oil bath because LiBH4as the solid electrolyte behaves a superior ionic conductivity at this temperature.22) And the capacity unit would be calculated due to the gravimetric amount of VH2. To investigate structural change of the electrode material, X-ray diffraction (XRD) measurements were performed by using Cu-K¡ radiation at room temper-ature (RINT-2500V, Rigaku Co.). All the processes of the above sample synthesis, assembly/disassembly of the coin cell, and the preparation of samples for the XRD measure-ments were carried out in a glove boxfilled with purified Ar (99.9%, 1 MPa).
3. Results and Discussion
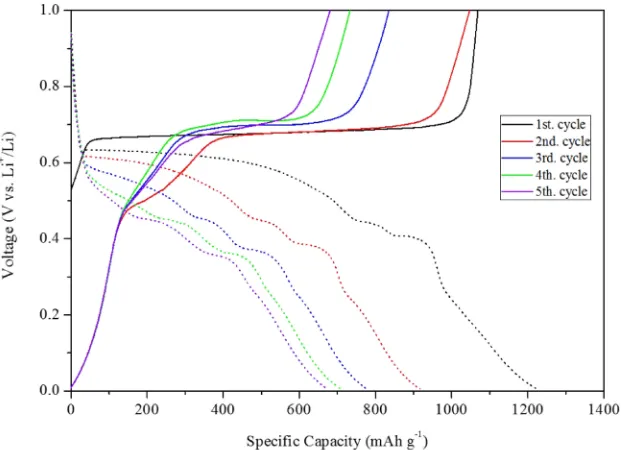
Figure 1 shows charge/discharge profiles of the V-LiH electrode for the 5 cycles. Because VH2 is unstable without hydrogen pressure, the initial state of 1st cycle is V+3LiH and the charging properties are examined atfirst. As is shown in Fig. 1, the charging process are only started from 0.53 V. However, the other cycles for charging are started from 0 V. For the XRD profile corresponding to 0.53 V (start), not only V but also VH0.81 are confirmed although the starting materials are V and LiH. This phenomenon indicates that a reaction of V+0.81LiH¼VH0.81+0.81Li partially pro-ceeds as a solid-solid reaction during mechanical ball-milling process before the electrochemical test. And, the plateau voltage around 0.65 V for the 1st charging should be corresponding to VH0.81+1.19LiH¼VH2+1.19Li. How-ever, the amount of 1.19Li has only ca. 600 mAh/g capacity. The capacity of 600 mAh/g obtained for the 1st charging is corresponding to the length of plateau, which is much smaller than that of total capacity of 1069 mAh/g. The extra 469 mAh/g is due to de-lithiation from lithiated carbon which might be generated during the mechanical ball-milling process from the reaction between acetylene black and Li
Fig. 1 Galvanostatic charge-discharge curves for V-LiH electrode in voltage range of 0.005 V1.0 V at 125°C. Y. Matsumura, K. Takagishi, H. Miyaoka and T. Ichikawa
[image:2.595.144.455.545.770.2]mentioned above. And then, electrochemical reaction for the 1st charging process shows conversion process to generate VH2 for the plateau region and de-lithiation process from CLix for the slope region. However, as shown in Fig. 2
corresponding to 1.0 V, the generated product is VH0.81 although the expected product should be VH2, indicating that the generated product does not change even after charging process with 600 mAh/g capacity. However, as mentioned above, VH2is unstable without H2 pressure. The generated VH2 must be decomposed to incomplete hydride phase VH0.81 with slightly high stability during the XRD measurement.
With respect to the 1st discharge process, about 1230 mAh/g capacity in total was shown with a prompt slope (10.6 V; ca. 30 mAh/g), a first plateau (0.60.45 V, ca. 720 mAh/g), and the other slope (0.450 V, ca. 480 mAh/g), indicating that the total capacity is larger than 1013 mAh/g corresponding to reaction (7). Because the expected coulombic efficiency corresponding to VH2 conversion reaction cannot be reached to 100%, this total capacity of 1230 should contain not only conversion process but also lithiation process to acetylene black (hard carbon). Of course, V single phase was confirmed in the 0 and 0.3 V XRD profiles of Fig. 2. Here, focusing on the 2nd to 5th discharging cycles, the profiles corresponding to more than
0.6 V and less than 0.3 V are quite similar, and we assume that the contribution of lithiation to the hard carbon is 470 mAh/ g, which is essentially equivalent to the graphitized carbon black so far reported,45) and this value doesn’t drastically change with the increasing cycles. The remaining part should be corresponding to conversion reaction.
Figure 3 shows the discharging profiles of each cycle, which are subtracted by the expected lithiation profile corresponding to acetylene black shown in the inset of Fig. 3. As shown in Fig. 3, three plateaus are clearly recognized in the early stages of cycles. And the lengths of the low plateaus around 0.350.4 V shown by horizontal arrows did not change with the increasing cycles, indicating about 150 mAh/g. However, the lengths corresponding to the high voltage region shown by vertical arrows were decreased drastically from 600 to 50 mAh/g for the 1st cycle and 5th cycle, respectively. It is noteworthy that the high and low plateau voltages are decreased with increasing cycles but middle plateau voltage was kept with even increasing cycles.
As the discharging processes included not only the conversion reaction of V-LiH to VH2 but also the lithiation reaction of acetylene black, the charging processes after 2nd cycle should include the de-lithiation reaction with 470 mAh/g capacity. Again, focusing on the 2nd cycle charging profile shown in Fig. 1, the slope regions below 0.45 V (130 mAh/g) and above 0.75 V (90 mAh/g) show about 220 mAh/g capacity, indicating that remaining capacity of 250 mAh/g should be hidden in the plateau regions between 0.45 and 0.65 V. Therefore, the remaining capacities of ca. 250 mAh/g corresponding to 0.45 V plateau and of ca. 580 mAh/g corresponding to 0.6 V plateau can be explained by the conversion reactions of V+0.81LiH¼
Fig. 2 Ex-situXRD patterns of V-LiH electrode at the various states of charging-discharging process. The profiles of 0.6 (start) and 1.0 V are corresponding to initial andfinal states for the 1st charging, respectively. The profiles of 0.5, 0.3 and 0.005 V are corresponding to each voltage for the 1st discharging.
[image:3.595.70.271.67.401.2] [image:3.595.308.545.71.320.2]VH0.81+0.81Li and VH0.81+1.19LiH¼VH2+1.19Li, respectively. And, with increasing cycles, the charging processes are reproduced well to have slimilar profiles in the conversion and lithiation reactions. Of course, the capacities corresponding to more than 0.5 V for the charging show the slightly decreased phenomena, indicating a good agreement with the decreasing trend of higher plateau region capacity of the 2nd to 5th discharging processes.
Finally, the capacity corresponding to the de/lithiation reaction of acetylene black would be discussed. The capacity of 470 mAh/g should be calculated due to the gravimetric amount of VH2. Therefore, the capacity calculated from the amount of carbon should be different. Actually, the amount acetylene black is 30/40 ratio compared with the amount of V+3LiH. Because the total molecular weight corre-sponding to V+3LiH is 75, the corresponding VH2 and carbon amounts should be 53 and 56, respectively. Therefore, 470 mAh/g capacity due to the VH2 weight should be converted to 444 mAh/g due to the carbon weight, indicating this value is quite reasonable as is assumed by lithiation of hard carbon.
4. Conclusion
In this study, we focused on the mechanism of V and LiH conversion reaction by using the electrochemical properties for all solid state LIBs. As a result of the electrochemical performance, the initial state of V and LiH should be partially converted to VH0.81during initial mixing by mechanical ball-milling. Of course, the by-product Li should be reacted with acetylene black, generating the lithiated hard carbon. Therefore, the 1st charging process shows slightly low capacity even though the capacity is decreased with increasing cycles. The 1st charging plateau at 0.65 V should be explained by the following conversion reaction, VH0.81+ 1.19LiH¼VH2+1.19Li.
In the over all charging/discharging processes, the stable and extra capacity of 470 mAh/g is included as de-lithiation/ lithiation processes, respectively. For the discharging processes after subtracted by hard carbon contribution, there are three plateau are obtained. Although the corresponding reaction to each conversion reaction are not clear, high plateau region shown between 0.5 and 0.6 V should be explained by the opposite reaction mentioned above, in which the capacity is drastically decreased with increasing cycles. On the other hand, the capacity corresponding to middle and low plateau regions shown around 0.4 V is not changed with even increasing cycles. Finally, in the charging processes, the plateaus of 0.5 and 0.6 V are, respectively, corresponding to the conversion reactions to generate VH0.81 and VH2, and as extra capacity corresponding to the slope regions of charging process, the de-lithiation reaction proceeded.
Acknowledgements
The authors acknowledge thefinancial support of the JSPS SAKURA Program.
REFERENCES
1) J.W. Kim, J.H. Ryu, K.T. Lee and S.M. Oh:J. Power Sources147
(2005) 227233.
2) U. Kasavajjula, C. Wang and A.J. Appleby: J. Power Sources163
(2007) 10031039.
3) W.J. Weydanz, M. Wohlfahrt-Mehrens and R.A. Huggins: J. Power Sources8182(1999) 237242.
4) Y. Yao, M.T. McDowell, I. Ryu, H. Wu, N. Liu, L. Hu, W.D. Nix and Y. Cui:Nano Lett.11(2011) 29492954.
5) Y. Oumellal, A. Rougier, M. Tarascon and L. Aymard: J. Power Sources192(2009) 698702.
6) S. Brutti, G. Mulas, E. Piciollo, S. Panero and P. Reale:J. Mater. Chem.
22(2012) 1453114537.
7) W. Zaïdi, J.P. Bonnet, J. Zhang, F. Cuevas, M. Latroche, S. Couillaud, J.L. Bobet, M.T. Sougrati, J.C. Jumas and L. Aymard:Int. J. Hydrogen Energ.38(2013) 47984808.
8) Y. Oumellal, A. Rougier and L. Aymard:Int. J. Hydrogen Energ.37
(2012) 78317835.
9) J.X. Zhang, W. Zaidi, V. Paul-Boncour, K. Provost, A. Michalowicz, F. Cuevas, M. Latroche, S. Belin, J.P. Bonnet and L. Aymard:J. Mater. Chem. A1(2013) 47064717.
10) S. Sartori, F. Cuevas and M. Latroche:Appl. Phys. A122(2016) 135.
11) Y. Oumellal, A. Rougier, G.A. Nazri, J.M. Tarascon and L. Aymard:
Nat. Mater.7(2008) 916921.
12) X. Yu, J.B. Bates, G.E. Jellison, Jr. and F.X. Hart:J. Electrochem. Soc.
144(1997) 524532.
13) R. Mercier, J.P. Malugani, B. Fahys and G. Robert:Solid State Ionics5
(1981) 663666.
14) A. Pradel and M. Ribes:Mater. Chem. Phys.23(1989) 121142.
15) M. Tatsumisago, K. Hirai, T. Minami, K. Takada and S. Kondo:
J. Ceram. Soc.101(1993) 13151317.
16) K. Takada, N. Aotani, K. Iwamoto and S. Kondo:Solid State Ionics
8688(1996) 877882.
17) R. Kanno and M. Murayama:J. Electrochem. Soc.148(2001) A742 A746.
18) M. Murayama, R. Kanno, M. Irie, S. Ito, T. Hata, N. Sonoyama and Y. Kawamoto:J. Solid State Chem.168(2002) 140148.
19) M. Murayama, N. Sonoyama, A. Yamada and R. Kanno:Solid State Ionics170(2004) 173180.
20) T. Kobayashi, A. Yamada and R. Kanno:Electrochim. Acta53(2008) 50455050.
21) T. Kobayashi, Y. Imade, D. Shishihara, K. Honma, M. Nagao, R. Watanabe, T. Yokoi, A. Yamada, R. Kanno and T. Tatsumi:J. Power Sources182(2008) 621625.
22) M. Matsuo and S. Orimo:Adv. Energy Mater.1(2011) 161172.
23) M. Matsuo, Y. Nakamori, S. Orimo, H. Maekawa and H. Takamura:
Appl. Phys. Lett.91(2007) 224103.
24) A. Unemoto, M. Matsuo and S. Orimo:Adv. Funct. Mater.24(2014) 22672279.
25) A. Unemoto, S. Yasaku, G. Nogami, M. Tazawa, M. Taniguchi, M. Matsuo, T. Ikeshoji and S. Orimo: Appl. Phys. Lett. 105 (2014) 083901.
26) K. Takahashi, K. Hattori, T. Yamazaki, K. Takada, M. Matsuo, S. Orimo, H. Maekawa and H. Takamura:J. Power Sources226(2013) 6164.
27) A. Unemoto, K. Yoshida, T. Ikeshoji and S. Orimo:Mater. Trans.57
(2016) 16391644.
28) Y. Zhou, M. Matsuo, Y. Miura, H. Takamura, H. Maekawa, A. Remhof, A. Borgschulte, A. Züttel, T. Otomo and S. Orimo:Mater. Trans.52
(2011) 654657.
29) P. López-Aranguren, N. Berti, A.H. Dao, J. Zhang, F. Cuevas, M. Latroche and C. Jordy:J. Power Sources357(2017) 5660.
30) L. Zeng, K. Kawahito, S. Ikeda, T. Ichikawa, H. Miyaoka and Y. Kojima:Chem. Commun.51(2015) 97739776.
31) L. Zeng, T. Ichikawa, K. Kawahito, H. Miyaoka and Y. Kojima:ACS Appl. Mater. Interfaces9(2017) 22612266.
32) K. Kawahito, L. Zeng, T. Ichikawa, H. Miyaoka and Y. Kojima:Mater. Trans.57(2016) 755757.
33) J.J. Reilly and R.H. Wiswall:Inorg. Chem.9(1970) 1678.
34) G.G. Libowitz and A.J. Maeland:J. Less-Comm. Met.131(1987) 275
282.
35) H. Yukawa, D. Yamashita, S. Ito, M. Morinaga and S. Yamaguchi:
J. Alloy. Compd.356357(2003) 4549.
36) K. Fujita, Y.C. Huang and M. Tada:J. Japan Inst. Metals43(1979) 601610.
37) M.V. Lototsky, V.A. Yartys and I.Yu. Zavaliy:J. Alloy. Compd.404 406(2005) 421426.
38) H. Yukawa, M. Takagi, A. Teshima and M. Morinaga:J. Alloy. Compd.
330332(2002) 105109.
39) H. Yukawa, A. Teshima, D. Yamashita, S. Ito, S. Yamaguchi and M. Morinaga:J. Alloy. Compd.337(2002) 264268.
40) H. Yukawa, D. Yamashita, S. Ito, M. Morinaga and S. Yamaguchi:
Mater. Trans.43(2002) 27572762.
41) S. Ito, D. Yamashita, K. Komiya, H. Yukawa and M. Morinaga:
J. Alloy. Compd.364(2004) 137140.
42) R. Kawai, H. Yukawa, A. Suzuki, T. Nambu and Y. Murata:Int. J. Hydrogen Energ.42(2017) 2256422574.
43) E. Veleckis and R.K. Edwards:J. Phys. Chem.73(1969) 683692.
44) S. Kumar, A. Jain, T. Ichikawa, Y. Kojima and G.K. Dey: Renew. Sustain. Energy Rev.72(2017) 791800.