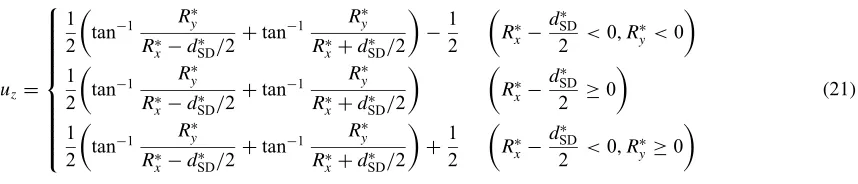Analysis of Dislocation Core Structure in B2 Ordered Phase
by Cluster Variation Method
Yasunori Yamada
1,+and Tetsuo Mohri
1,21Division of Materials Science and Engineering, Graduate School of Engineering, Hokkaido University, Sapporo 060-8628, Japan 2Research Center for Integrative Mathematics, Hokkaido University, Sapporo 060-8628, Japan
Theoretical framework of the Cluster Variation Method (CVM) is extended in two directions. One is the construction of a supercell in which the basic clusters are aligned in two dimensional directions. The other one is the introduction of the atomic displacement in theflexible lattice. These extensions of the conventional CVM enable us to calculate a core structure of two parallel superpartial dislocations in B2 ordered phase atfinite temperatures. It is shown that a large stacking fault is formed inside the two superpartial dislocations at lower temperatures, while the stacking fault appears outside the superpartial dislocations at higher temperatures. [doi:10.2320/matertrans.MAW201214]
(Received April 25, 2012; Accepted June 18, 2012; Published August 1, 2012)
Keywords: cluster variation method, core structure of dislocation, anti-phase boundary, B2 ordered phase
1. Introduction
A number of ordered intermetallic compounds exhibit a positive yield stress dependence at elevated temperatures, which is known as the yield stress anomaly, and various mechanisms have been proposed to explain the anomaly. Among them are KearWilsdorf locks,1,2)order strengthening
model,3) climb dissociation model4)and vacancy hardening
model.5)The detailed mechanism which can be applied to B2
ordered compound, however, is not completely clear. One of the potential approaches to clarify the yield stress anomaly is to analyze the dislocation core structure. In 1970, the calculation for a core structure of a 1/2 h111i screw dislocation in bcc crystals was performed by Vitek, et al.6)
by using the central-force potential. In this study, they analyzed the core structure with a displacement map, known as Vitek map, and they predicted that the core structure possessed three-fold symmetry. Recently, Mori
et al. investigated the core structure of a dislocation and
Peierls stress in a bcc metal based on microscopic
phase-field model,7) and they confirmed that the Peierls stress of a 1/2 h111i screw dislocation in bcc Fe with non-planar structure was appreciably higher than that with planar structure. These results imply the importance of the core structure for the study of yield stress anomaly. Besides these, there are other potential approaches such as the fi rst-principles calculation8) and PeierlsNabarro method.9) The
applicability of these studies, however, is limited to 0 K, since the entropy effect is not explicitly taken into considerations and the temperature dependence is by no means considered. Furthermore, it is pointed out that Anti Phase Boundary (hereafter abbreviated as APB) plays a significant role for a yield stress anomaly in ordered intermetallic compounds,14)which is again expected to be dependent on temperature. Therefore, a new comprehensive approach which is able to deal with the core structure including APB at finite temperatures is required for more satisfactory studies on yield stress anomaly.
Cluster Variation method (hereafter abbreviated as CVM) devised by Kikuchi10)has been regarded as one of the most reliable theoretical tools to study atomistic arrangements in the equilibrium state of an alloy system. CVM provides a series of approximated entropy expressions in terms of cluster probabilities of a basic cluster and its sub-clusters. The level of the approximation is determined by the size of the basic cluster which is the biggest cluster contained in the entropy formula, and the bigger the basic cluster is the better results one can expect although the computational burden becomes heavier.
CVM has been mainly employed to calculate phase diagrams, and it has been confirmed that the topology of the phase boundaries and the order of the transitions are correctly predicted. Besides these, the optimized correlation functions and atomic separation provide temperature and concentration dependences of order parameters and equi-librium lattice constant.11,12)
Another application of the CVM is the study of atomic arrangement at an interface. For such a calculation, a supercell extended perpendicular to the interface is con-structed as the succession of a basic cluster. Kikuchi13)
employed such a supercell and calculated the width of (110) boundary in bcc structure within the Ising model. Then, Asta14) extended the procedure to a more realistic
alloy by introducing the Effective Cluster Interaction (ECI) parameters, and he calculated the profiles of composition and order parameters as well as the interfacial energy of the coherent interphase boundary between¡and¤Aphase in ALLi alloy.
These calculations motivate us to align basic clusters in two dimensions so that more extended defect structures can be studied. One of these defects is a dislocation of which atomistic structure is basically the stacking of a two dimensional symmetric structure along the extension of the dislocation line. However, the description of dislocation core requires the calculation of the relative displacement of atoms, which is not considered by conventional CVM. Accordingly, in order to introduce the relative displacement of atoms into CVM we employed flexible lattice and +Graduate Student, Hokkaido University
calculated equilibrium lattice positions by minimizing the free energy. Thereby, we proposed the comprehensive calculation model for the dislocation core structure including APB atfinite temperatures, and analyzed the core structure of two parallel superpartial dislocations in B2 ordered phase.
2. Calculation Procedure
2.1 Free energy model
Formulation of the entropy is the core of the present study and the details are provided in the following.
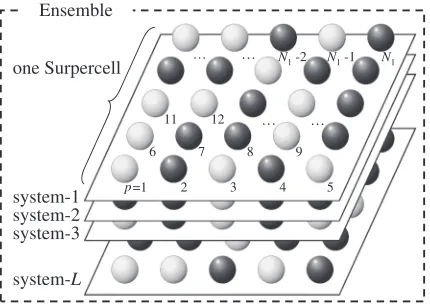
We consider an ensemble of supercell systems which is shown in Fig. 1. Here, the supercell is defined as the collection of atoms (point clusters). The possible number of systems, WL, in the ensemble is given by the following
equation within the pair approximation of CVM.
WL¼
Y
p
Wptp
Y
p;q
Gpqpair ð1Þ
whereWptp is the number of possible ways of arranging atoms on the column of the lattice pointpandGpqpair is a correlation correction factor between p-point and q-point. TheWptp and Gpqpair are written as.15)
Wptp ¼Y L!
i
ðLxpiÞ! ð2Þ
and
Gpqpair¼
Y
i
ðLxpiÞ!Y
j
ðLxqjÞ!
Y
i;j
ðLypqijÞ!L! ð3Þ
When the ensemble is constructed as an arrangement ofN
supercells,WL is given as:
WL ¼
Y
n
Y
p;q
Wptp,n
Y
n;m
Y
p;q
Gpq,nmpair ð4Þ
where n(m) specifies the supercell and Gpq,nmpair is the correlation correction factor for the pair between p-point in the n-th supercell and q-point in the m-th supercell. By substituting eqs. (2) and (3) into eq. (4), we obtain the entropy per supercell by employing Stirling’s approximation as:
S
N ¼kBlnW¼
kB
L lnWL
¼ kB N
" X
p;n
X
i
xp,ni lnxp,ni þ X
p;q;n;m
X
i;j
ypq,nmij lnypq,nmij
X
i
xp,ni lnxp,mi X
j
xq,mj lnxq,mj
#
ð5Þ
where kB is the Boltzmann constant. It is further noted that
when the net interaction betweenij pair is absent, the pair probabilityypqij satisfies the following relation,
ypq,nmij ¼xp,ni xq,mj ð6Þ
and, therefore, one reaches the relation written as,
X
i;j
ypq,nmij lnypq,nmij X
i
xp,ni lnxp,ni
X
j
xq,mj lnxq,mj ¼0 ð7Þ
Then, for the summation over p and q in eq. (5), it is only necessary to consider the pair clusters which are interacting each other. We defineZppair as the number of such interacting pair clusters which involve the p point, and the set of interacting pair cluster in the system is denoted as A. The entropy per supercell is, therefore, rewritten in a more tractable form as
S
N ¼
kB
N
" X
p;n
X
i
ð1ZppairÞxp,ni lnxp,ni
þ X
fp;q;n;mg2A
X
i;j
ypq,nmij lnypq,nmij
#
ð8Þ
By noting that the cluster probabilities are equivalent in all the supercells, xp,ni and ypq,nmij can be reduced to xpi and ypq,ij k, respectively, where k specifies the relative position between the n- andm-th supercell. Then, eq. (8) is further rewritten as
S
N ¼ kB
" X
p
X
i
ð1ZppairÞxpi lnxpi
þ X
fp;q;kg2A
X
i;j
ypqij;klnypqij;k
#
ð9Þ
In this way, the entropy formula is constructed for the present study. It is emphasized that this entropy formula can be applied to any crystal symmetry of the supercell including a dislocation core in the present study.
The internal energy,E, in the present study is formulated within the pair interaction model and is given by
E
N ¼
X
fp;q;kg2A
X
i;j
eijðrpq;kÞypqij;k ð10Þ
and
rpq;k¼ kRp,nRq,mk ð11Þ
whereeij(rpq,k) is the pair interaction energy between atomic
species i and j located at the lattice points p and q, respectively, rpq,k is the interatomic distance between the
lattice pointspand q, andRp,n(Rq,m) is a position vector of the lattice point p(q).
Ensemble
system-L system-3 system-2 system-1 one Surpercell
p=1 2
… N1 -2 N1 -1 N1
11 …
6
12
5 4 3
9 8 7
… …
[image:2.595.63.278.71.224.2]By combining eqs. (9) and (10), the grand potential per supercell is given as
N ¼
X
fp;q;kg2A
X
i;j
eijðrpq;kÞypqij;k
þkBT
" X
p
ð1ZppairÞX
i
xpi lnxpi
þ X
fp;q;kg2A
X
i;j
ypqij;klnypqij;k
#
X
p
X
i
®ixpi ð12Þ
where®iis the chemical potential of the speciesi. It should
be noted that, in eq. (12),fxpigandfypq,ijkgare not independent variables and must satisfy the following normalization and geometrical conditions,
X
i
xpi ¼1 and X
i;j
ypqij;k¼1 ð13Þ
and
X
j
ypqij;k¼xpi ð14Þ
In order to avoid numerical complicacies, it is more desirable to introduce a set of independent variables known as correlation functions.
In fact, it has been amply demonstrated15)that the cluster probabilities xpi and ypq,ijk are related to point and pair correlation functionsf²p1gand f²pq2;kgthrough
xpi ¼1
2ð1þi²
p
1Þ ð15Þ
and
ypqij;k¼1
4ð1þi²
p
1þj²q1þij²pq2 ;kÞ ð16Þ
where,itakes+1(¹1) whenpis occupied by A(B) atom. A similar definition is applied to j and the occupation at q. Substitution of eqs. (15) and (16) into eq. (12) yields the free energy function as:
N ¼
X
fp;q;kg2A
X
i;j
1
4eijðr
pq;kÞð1þi²p
1þj²q1þij²pq2;kÞ
þkBT
X
p
ð1ZppairÞX
i
1
2ð1þi²
p
1Þlnð1þi²p1Þ
þ X
fp;q;kg2A
X
i;j
1
4ð1þi²
p 1þj²
q 1þij²
pq;k
2 Þlnð1þi² p 1þj²
q 1þij²
pq;k
2 Þ
2 6 6 6 4
3 7 7 7 5
X
p
X
i
®i1
2ð1þi²
p
1Þ ð17Þ
2.2 Minimization
Given T, µi and a supercell, that is fZppairg, the cluster
probabilities in equilibrium state are obtained by minimizing eq. (17). For a flexible lattice, the independent variables in the present study are the correlation functionsf²p1g,f²pq2 ;kgand the position vector {Rp}. Then, these variables satisfy the
simultaneous equations at the equilibrium state,
rf¼ @
@f²p1g;
@
@f²pq2;kg;
@
@fRpg
!
¼0 ð18Þ
By solving eq. (18) with Conjugate Gradient method,16,17) the equilibrium state is determined.
2.3 Calculation condition
In the present study, we employed central-force potential which was proposed by Vitek6) and is expressed in the
following polynomial form:
eijðrpq;kÞ ¼
X5
n¼0
bij;nðrpq;kÞn ð19Þ
where bij,n is a phenomenological constant. The interaction
has been considered up to second-nearest neighbors, which is necessary to stabilize a bcc structure by using solely central force. To ensure the stability of a B2 ordered phase at low temperatures, a deeper potential is assigned to unlikeatomic pairs. Moreover, the equilibrium atomic distance for the unlike pair is set to be smaller than those for the likepairs. The interaction energies and the nondimensional parameter
bij,nemployed for the present study are shown in Fig. 2 and
Table 1, respectively.
It is noted that the energy and length scales are normalized with respect to the bottom value of the interaction energy,
e0
AA, between A-atoms. Followings are the normalized parameters employed in the present study,
Rp ¼Rp=r0 AA; rpq
¼rpq=r0AA;
aeq¼aeq=r0AA; T
¼
kBT =e011
eij¼eij=e0AA; b
ij;n¼bij;n=ðe0AA=r0AA nÞ
;
APB¼APB=ðe011=r0AA2Þ; ®i ¼®i=e0AA ð20Þ
where aeq is an equilibrium lattice constant andAPB is an
Anti Phase Boundary energy.
-2.0 -1.5 -1.0 -0.5 0.0 0.5
atomic interaction ene
gy ,
eij
*
interatomic distance, rpq* A-A, B-B A-B )
,
(rAA0 e0AA
0.8
−
1.4 1.3 1.2 1.1 1.0 0.9
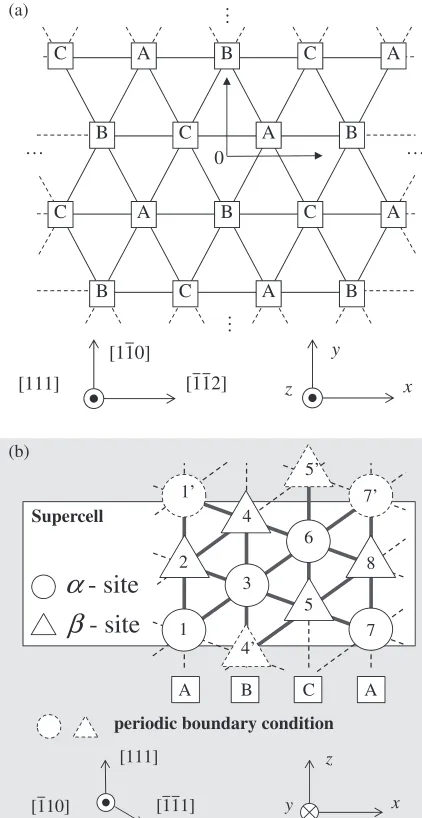
[image:3.595.57.289.99.203.2]To calculate the core structure of the dislocation, we used the supercell in which the point and pair clusters are arranged in the two dimensional plane which is perpendicular to the dislocation line as shown in Figs. 3(a) and 3(b). In these
figures, x, y and z-directions correspond to [112], [110]
and [111], respectively. When a pair of 1/2[111] screw dislocations are introduced on (110) plane, the initial atomic displacements in z-direction, uz, are given by the following
equations
uz¼
1
2 tan
1 R
y
RxdSD=2þtan
1 R
y
RxþdSD =2
1 2 R x dSD
2 <0; R
y<0
1 2 tan 1 R y
RxdSD=2þtan
1 R
y
RxþdSD =2
Rxd
SD
2 0
1 2 tan 1 R y
RxdSD=2þtan
1 R
y
RxþdSD =2
þ1 2 R x dSD
2 <0; R
y0
8 > > > > > > > > < > > > > > > > > :
ð21Þ
wheredSD is the distance between the two dislocations with dSD¼8pffiffiffi6=3aeq. We used a discus-shaped supercell whose radius and thickness in thez-direction are20pffiffiffi6=3 aeq and 2pffiffiffi6=3aeq, respectively. Periodic boundary condition is imposed along the axis direction while the free boundary condition is employed in the radial direction. In this manner, we deal with the dislocation core within the CVM.
The pair clusters are sought by an algorithm developed for the present study and pairs of which distance are shorter than
some critical value are identified. As the critical value we assign 1:95pffiffiffi6=3aeq which is long enough to contain all the interacting pair clusters during the lattice relaxation process. This search is conducted only at the initial step of the calculation.
The equilibrium lattice constant aeq is determined by minimizing the free energy of the bulk phase which was given by eq. (12) with the substitution of 8 and 6 for the nearest and next nearest neighbor pairs, respectively,
N ¼4
X
i;j
eijðr¡¢Þy¡¢ij þ
3 2
X
i;j
eijðr¡¡ÞYij¡¡þ
3 2
X
i;j
eijðr¢¢ÞYij¢¢
þkBT
13
2
X
i
x¡i lnx¡i 13
2
X
i
x¢i lnx¢i
þ4X
i;j
y¡¢ij lny¡¢ij þ3
2
X
i;j
Yij¡¡lnYij¡¡þ3
2
X
i;j
Yij¢¢lnYij¢¢
2 6 6 6 4 3 7 7 7 5 1 2 X i
®ix¡i
1 2
X
i
®ix¢i ð22Þ
wherex¡i andx¢i are the point probability at¡and¢sublattice sites for a B2 ordered phase, respectively, and y¡¢ij and Yij¡¡ (orYij¢¢) are pair probabilities for thefirst and second nearest neighbors, respectively.
In order to analyze the atomic arrangement of the dislocation core structure, the following Long Range Order parameter (here after LRO)SB2in [111] direction is defined,
SB2¼x¢
0
B x¡
0
B ð23Þ
wherex¡B0 andx¢B0 are the point probability offinding B atom at¡and¢sublattice sites which are located on the same line normal to (111) plane.
Finally, the chemical potential®iis set to null, and for this
condition the equilibrium average concentration xB is automatically kept to bexB¼0:5.
3. Results and Discussions
First, we calculate the equilibrium LRO and lattice constant in the bulk phase by minimizing eq. (22). The results are demonstrated in Fig. 4.
[image:4.595.121.550.144.236.2]One sees that the LRO decreases smoothly and monotoni-cally with increasing the temperature towards T*=2.30 at which the LRO vanishes. Hence, T*=2.30 is identified as the transition temperature. Also, from the smooth behavior of the LRO curve, one may conclude that the order of the phase Table 1 Calculation parameter for atomic interaction in eq. (19).
eAA,eBB eAB
a1 0.93846154 0.891538
a2 1.11538462 1.059615
a3 1.32307692 1.256923077
eAA,eBB
rpq <a
1 a1<rpq
<a2 a2<rpq
<a3 a3<rpq
b0 1447.335512 23448.68922 1398.11666 0 b1 ¹6468.243513 ¹112289.4168 ¹4616.759013 0 b2 10993.88088 214858.162 5678.461616 0 b3 ¹8395.790925 ¹205328.7024 ¹3086.300991 0 b4 2422.244163 97992.13957 625.9136002 0
b5 0 ¹18681.87169 0 0
eAB,eBA rpq <a
1 a1<rpq
<a2 a2<rpq
<a3 a3<rpq
b0 2171.003 35173.03 2097.17499 0 b1 ¹10213 ¹177299 ¹7289.619495 0 b2 18272.38 357105 9437.886342 0 b3 ¹14688.7 ¹359228 ¹5399.564353 0 b4 4460.821 180463 1152.686551 0
transitions is of the second-order. The equilibrium lattice constant increases with the temperature. This is purely a configurational effect, namely the number of like pairs, for which we assigned the larger atomic distance than unlike
pairs as shown in Fig. 2, increases with temperature. One may also recognize a discontinuous change of the lattice constant due to the transition atT*=2.30. The equilibrium lattice constants determined at a given temperature in Fig. 4 is utilized in the later calculation.
LRO and atomic displacement in the system which includes a pair of parallel 1/2[111] screw dislocations connected by APB are calculated by minimizing eq. (17) with cluster probabilities and lattice positions. The results are shown in Figs. 5(a), 5(b) and 5(c), where each sphere indicates a lattice point and the color scale displays the absolute value of LRO. The APB and the dislocations are
[111]
[110]
[112] z
y
x 0
A B C A
C
A B A
C C
A
B C B
A
B C B
… …
…
…
(a)
Supercell
- site
α
- site
β
periodic boundary condition
[110]
[111]
[111] y
z
x
(b)
A C B A
7 8 7’
5 6 5’
4’ 3 4
1 2 1’
Fig. 3 The alignment of sublattice points in supercell. (a) the projection of the bcc (111) plane (b) (110) plane.
1.06 1.065 1.07 1.075 1.08
0 0.2 0.4 0.6 0.8 1
0
Lattice constant,
aeq
*
LR
O,
SB2
Temperature, T*
LRO
Lattice constant
3 2
1
Fig. 4 The temperature dependences of the equilibrium LRO and lattice constant. The lattice constant is normalized with respect to the interatomic distance corresponding to the minimum of the atomic interaction energy between A-atoms.
[111] [112]
[110]
Screw Dislocation ( b=1/2[111] )
APB (b)
[111] [112]
[110]
Screw Dislocation ( b=1/2[111] )
APB (a)
Absolute v
a
lue of LR
O,
||B2
|
S
Absolute v
alue
of
LR
O,
|
SB2
|
Stacking Fault
[111] [112]
[110]
Screw Dislocation ( b=1/2[111] )
APB (c)
Absolute v
alue of LR
O,
|
SB2
|
[image:5.595.313.541.65.600.2] [image:5.595.63.274.71.480.2] [image:5.595.68.272.533.673.2]extending in the normal direction to the figure, and the two dislocations are shown as double circles. One sees that the higher the temperature is, the lower the LRO becomes. The LRO in the bulk phase at each temperature is confirmed to be the same as shown in Fig. 4. AtT*=1.10, the LRO is nearly 1.0 in the entire system, indicating the perfect ordered state even around dislocations and APB. However, at T*=1.50 and 2.10, the LRO in APB area obviously becomes lower than that in the bulk phase, and the thickness of the APB area increases with temperature. These configurations around APB at T*=1.50 and 2.10 are regarded as a relaxed structure. Furthermore, at T*=2.10, the area with lower LRO appears outside the APB area, where atomic planes are displaced which disturbs the regular bcc structure, resulting in the formation of a stacking fault, and the details are discussed later.
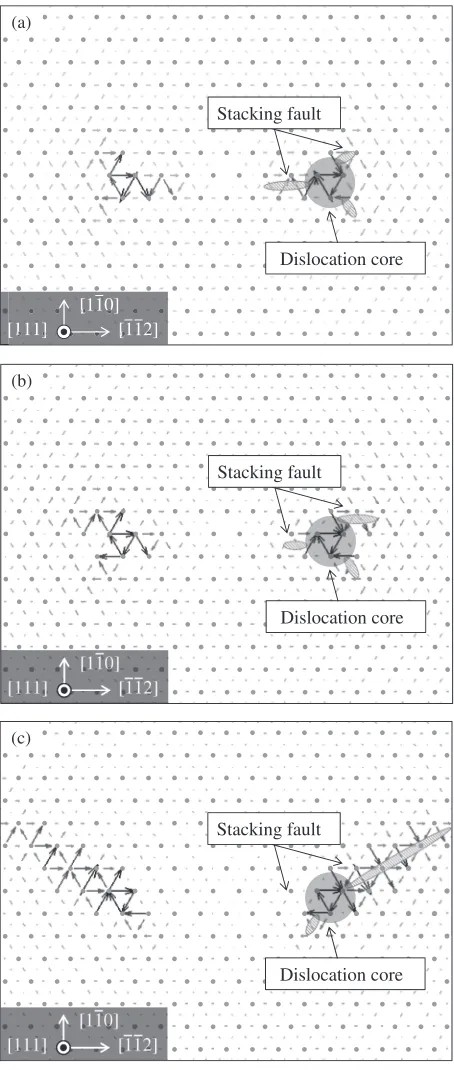
In order to analyze the core structure of the pair of two screw dislocations, displacement maps proposed by Vitek
et al.6)is employed as shown in Figs. 6(a), 6(b) and 6(c). In
thefigure, the dots show the projection of the lattice points on (111) plane and the arrows between the two dots indicate the difference of the atomic displacement in [111] direction from a perfect bcc crystal. A triangle of the arrows forming a cycle indicates the position of the dislocation core, and the planes in which arrows are directed in certain directions can be regarded as a stacking fault. However, it should be noted that the unambiguous separation of the dislocation cores and stacking faults is generally a difficult task.
In Figs. 6(a) and 6(b) at T*=1.10 and 1.50, the dislocations core posses the three-fold symmetry structure which confirms the result obtained by Vitek.6) However,
more careful observation suggests that the present results do not show the perfect three-fold symmetry and an alignment of the stacking faults also changes with temperature. At
T*=1.10 the larger stacking faults are formed inside two dislocations on{110}plane, by contrast, atT*=2.10 much larger stacking faults are extended outside two dislocations on {112} plane. The dislocation core at T*=1.50 is regarded as a transitional configuration between those at
T*=1.10 and 2.10. These changes of dislocation cores may be explained by the balance of the attractive and repulsive forces between the two dislocations. The two dislocations are subjected to attractive force in order to reduce the area of APB and the magnitude of the force is closely related to the APB energy. Then, the APB energy on {110} plane is calculated. The supercell is the same as that shown by Kikuchi13) and consist of fixed lattice, the free energy is
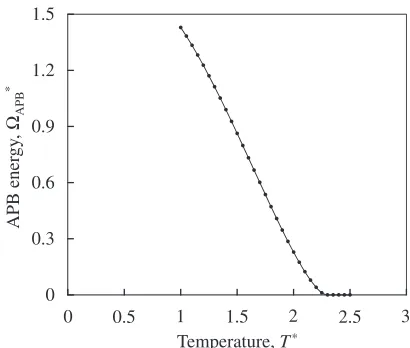
described by eq. (17) and the equilibrium lattice constants are shown in Fig. 4. The APB energy ³APB is obtained as the difference of the entire free energy with/without the APB
APB¼FAPBFbulk
AAPB
ð24Þ
where AAPB is the area of APB, FAPB and Fbulkare the free
energy of the entire system with/without APB, respectively. In Fig. 7, one sees that the APB energy decreases monotonically and vanishes at the temperature corresponding to the orderdisorder transition temperature,T*=2.30. This indicates that the attractive force between the two disloca-tions decreases with the increase of the temperature.
The repulsive force, on the other hand, is basically due to the elastic interaction acting between the two dislocations, and the magnitude of the force is determined by elastic module and the distance between the dislocations. When the distance between the dislocations is kept constant, it is considered that the repulsive force does not strongly depend on temperature because the temperature dependency of the elastic module is much smaller than that of APB energy. These considerations suggest that at low temperatures the two dislocations attract each other predominantly due to the effect of APB, while at high temperatures the two dislocations repel through the elastic interaction.
Dislocation core Stacking fault
[111] [112]
[110] (a)
Dislocation core Stacking fault
[111] [112]
[110] (b)
Dislocation core Stacking fault
[111] [112]
[110] (c)
[image:6.595.314.541.69.604.2]It is observed that in Fig. 6 the dislocations do not move despite the presence of attractive and/or repulsive forces due to the APB and the elastic interaction. This is because these forces are not sufficient enough to overcome the Peierls stress, hence the dislocation pairs are regarded in a transi-tional configuration. When a dislocation with non-planar core structure is subjected to forces, the core polarized on different planes is shrunk to form a planar structure on a single slip plane. This enables glide motion of a dislocation. Therefore, at T*=1.10, the core of dislocation is shrunk onto {110} and pair of dislocations move inward on the{110}plane due to the attractive interaction force originating from APB. This results in the formation of the larger stacking faults between the two dislocations. At T*=2.10, on the other hand, the core of dislocation is shrunk on the {112} followed by the outward motion of pair of dislocations driven by the elastic repulsive interaction. This is a mechanism of the formation of stacking faults outside of the pair of dislocations found in Fig. 6(c).
These results indicate that the core structure of dislocations in an ordered phase at finite temperature can be investigated by CVM, and two opposite mechanisms, attraction by APB and repulsion by the elastic interaction, explains the peculiar behavior of the dislocation motion in the ordered phase.
4. Conclusion
In this study, we extended the applicability of the Cluster Variation Method by using supercell in which the basic clusters are aligned in two dimensional directions. The entropy is formulated for irregular lattice and the resultant free energy is minimized with respect to cluster probabilities and lattice position. Such a new scheme of the CVM is
applied to study core structures of superpartial dislocations in B2 ordered phase atfinite temperatures. The main results are summarized as follows.
(1) AtT*=1.10, almost perfect ordered structure appeared in the whole system. However, atT*=1.50 and 2.10, the LRO in APB area were obviously lower than that in the bulk phase, and the formation of the APB at
T*=1.50 and 2.10 is regarded as the relaxed structure. (2) It was attempted to analyze the core structure of dislocation by displacement map proposed by Vitek.6) The dislocation core was characterized basically by the three-fold symmetry. The alignments of the stacking faults changed with temperature and these changes were explained by the balance between attractive force due to APB and repulsive force due to the elastic interaction.
Acknowledgments
This research was supported by Japan Science and Technology Agency (JST) under Collaborative Research Based on Industrial Demand “Heterogeneous Structure Control: Towards Innovative Development of Metallic Structural Materials”.
REFERENCES
1) B. H. Kear and H. G. F. Wilsdorf: Trans. Met. Soc. AIME224(1962) 382386.
2) V. Paidar, D. P. Pope and V. Vitek:Acta Metall.32(1984) 435448. 3) N. S. Stoloff and R. G. Davies:Acta Metall.12(1964) 473485. 4) Y. M. Zhu and H. Saka:Phil. Mag. A59(1989) 661676. 5) E. P. George and I. Barker:Phil. Mag. A77(1998) 737750. 6) V. Vitek, R. C. Perrin and D. K. Bowen:Phil. Mag.21(1970) 1049
1073.
7) H. Mori, H. Kimizuka and S. Ogata:J. Japan Inst. Metals75(2011) 104109.
8) F. Shimizu, S. Ogata, H. Kimizuka, T. Kano, J. Li and H. Kaburaki: J. Earth Simulator7(2007) 1721.
9) Q. Chen, M. Ji, C. Z. Wang, K. M. Ho and S. B. Biner:Intermetallics 18(2010) 312318.
10) R. Kikuchi:Phys. Rev.81(1951) 9881003.
11) J. M. Sanchez, J. R. Barefoot, R. N. Jarrett and J. K. Tien:Acta Metall. 32(1984) 15191525.
12) M. Enomoto and H. Harada:Metall. Mater. Trans. A20(1989) 649 664.
13) R. Kikuchi:J. Chem. Phys.66(1977) 33523362. 14) M. Asta:Acta Mater.44(1996) 41314136.
15) T. Mohri: Statistical Thermodynamics and Model Calculations,Alloy Physics, Chap. 10, ed. by W. Pfeiler, (Wiley-VCH, Weinheim, 2007) pp. 525588.
16) B. T. Polyak:USSR Comput. Math. Math. Phys.9(1969) 94112. 17) W. H. Press, B. P. Flannery, S. A. Teukolsky and W. T. Vetterling:
NUMERICAL RECIPES in C (Japanese Edition), (Gijutsu Hyoron Sha, Tokyo, 1993) pp. 282338 [in Japanese].
0 0.3 0.6 0.9 1.2 1.5
0
APB ener
gy
,
ΩAPB
*
Temperature, T*
3 2.5 2 1.5 1 0.5
[image:7.595.66.271.69.243.2]