Acta Crystallographica Section E
Structure Reports Online
ISSN 1600-5368
Chlorobis(
g
5-cyclopentadienyl)[
N
-(2,6-diisopropyl-phenyl)-
N
-(1-phenylvinyl)amide]zirconium(IV)
Benjamin L. Rupert,* John Arnold and Alexander Krajete
College of Chemistry, University of California, Berkeley, CA 94720, USA
Correspondence e-mail: brupert@berkeley.edu
Key indicators
Single-crystal X-ray study
T= 161 K
Mean(C–C) = 0.008 A˚
Rfactor = 0.039
wRfactor = 0.039
Data-to-parameter ratio = 10.9
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
Received 23 January 2006 Accepted 27 March 2006
#2006 International Union of Crystallography
The title compound, [Zr(C5H5)2Cl(C20H24N)], is the second
example of an enamido zirconocene to be crystallographically characterized. The geometry about nitrogen is planar, but the orientation of the aryl and vinyl substituents indicates that the nitrogen lone pair is not conjugated with their -systems. These features are attributed to the steric bulk of the complex.
Comment
Despite recent advances in non-metallocene polymerization chemistry, group IV metallocenes still represent the most commercially important olefin polymerization catalysts. While a large number of catalysts of this type are already known, further modification of both the cyclopentadiene rings and the sacrificial supporting ligands may lead to improved catalyst performance and polymer properties. In particular, the sacri-ficial ligands are important for initialization kinetics and end group functionalization. While these substituents most commonly are halides or alkyls, the use of amido ligands is also possible.
This report details the structure of a very bulky enamido complex (I), which is only the second enamido zirconocene for which the solid state structure has been reported, the first being Cp*CpZr(NHtBu)[N(Ph)C(Ph) CH2] (Zuckerman et
al., 2000). This compound could possibly be a pre-catalyst for olefin polymerization, or a precursor for a cationicd0alkene complex after abstraction of the chloride ligand.
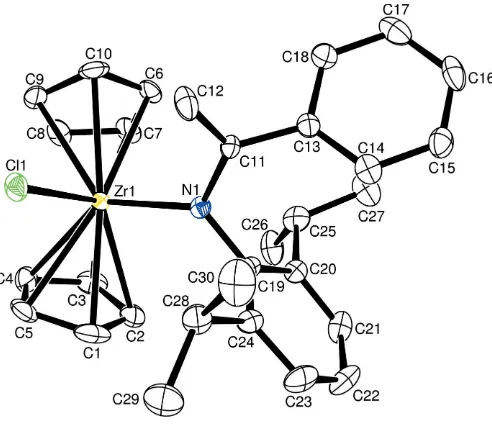
There are a few features of the structure (Fig. 1) that are worth mentioning. While the amido N atom has a perfectly planar configuration (sum of the bond angles around N1 is equal to 360.0), other structural features indicate that there is
87.5 (5)], precluding conjugation with this aromatic system as
well. Finally the Zr1—N1 bond distance is rather long at 2.141 (4) A˚ . More typical Zr—N bond lengths are in the range of 2.068 (3) A˚ found in Cp2Zr(Cl)N(PhCH2)2(Henderson et
al., 2002) to 2.096 (6) A˚ in Cp2Zr(Cl)NHPh (Grigsby et al.,
1996). The conformation of the enamido fragment and the long Zr—N bond are almost certainly a consequence of the bulky nature of the ligand.
Experimental
2.8M n-BuLi (3.6 mmol, 1.30 ml) in hexane was slowly addedviaa syringe to a cooled solution of diisopropylamine (0.51 ml, 3.6 mmol) in anhydrous tetrahydrofuran (THF; 20 ml). The cooling bath (213 K) was removed and the solution stirred for 1.5 h at room temperature. 2,6-Diisopropyl-N-(1-phenylethylidene)aniline (1.0 g, 3.6 mmol) (Kristen et al., 2001) was added at 213 K to the freshly prepared lithium diisopropylamide solution and the solution was stirred for 1.5 h. The solution was then transferred to a flask charged with zirconocene dichloride (1.0 g, 3.4 mmol) in anhydrous THF, which was cooled to 203 K. After stirring for 2 h at room temperature the solvent was removedin vacuoand the residue taken up in 40 ml toluene. The solution was filtered into a Schlenk tube and concen-trated. A yellow crystalline material (1.39 g, 72%) was obtained by cooling the solution to 278 K overnight. These crystals were used for the X-ray study without recrystallization.
Crystal data
[Zr(C5H5)2Cl(C20H24N)]
Mr= 535.28
Orthorhombic,P212121
a= 9.5134 (8) A˚
b= 14.718 (1) A˚
c= 18.591 (2) A˚
V= 2603.2 (4) A˚3
MoKradiation Cell parameters from 2777
reflections = 2.4–22.6
= 0.54 mm 1
T= 161.0 K Column, yellow
Data collection
Bruker SMART 1000 diffractometer !scans
Absorption correction: multi-scan (Blessing, 1995)
Tmin= 0.870,Tmax= 0.958
11464 measured reflections 4287 independent reflections 3249 reflections withI> 3(I)
Rint= 0.055 max= 24.7
Refinement
Refinement onF R[F2> 2(F2)] = 0.039
wR(F2) = 0.039
S= 1.09 3249 reflections 298 parameters
H-atom parameters constrained
w= 1/[2
(Fo) + 0.00022|Fo|2] (/)max= 0.001
max= 0.46 e A˚ 3 min= 0.68 e A˚ 3
Absolute structure: Flack (1983), 1807 Friedel pairs
[image:2.610.48.294.70.284.2]Flack parameter: 0.06 (5)
Table 1
Selected geometric parameters (A˚ ,).
Zr1—Cl1 2.471 (1)
Zr1—N1 2.141 (4)
N1—C11 1.443 (6)
N1—C19 1.461 (6)
C11—C12 1.340 (7)
Zr1—N1—C11—C12 38.4 (6) Zr1—N1—C19—C20 87.5 (5)
H atoms were positioned geometrically (C—H = 0.95 A˚ ) and refined as riding, withUiso(H) = 1.2Ueq(C).
Data collection:SMART(Bruker, 1999); cell refinement:SAINT (Bruker, 2002); data reduction:SAINT; program(s) used to solve structure:SIR92(Altomare et al., 1993); program(s) used to refine structure: TEXSAN (Molecular Structure Corporation & Rigaku Corporation, 1998); molecular graphics:ORTEP-3(Farrugia, 1997); software used to prepare material for publication:TEXSAN.
The authors thank Dr Frederick J. Hollander and Dr Allen G. Oliver for assistance and advice. The authors also thank the NSF for funding.
References
Altomare, A., Cascarano, G., Giacovazzo, C. & Guagliardi, A. (1993).J. Appl. Cryst.26, 343–350.
Blessing, R. H. (1995).Acta Cryst.A51, 33–38.
Bruker (1999).SMART. Version 5.054. Bruker AXS Inc., Madison, Wisconsin, USA.
Bruker (2002).SAINT. Version 6.40. Bruker AXS Inc., Madison, Wisconsin, USA.
Farrugia, L. J. (1997).J. Appl. Cryst.30, 565. Flack, H. D. (1983).Acta Cryst.A39, 876–881.
Grigsby, W. J., Olmstead, M. M. & Power, P. P. (1996).J. Organomet. Chem. 513, 173–180.
Henderson, K. W., Hind, A., Kennedy, A. R., McKeown, A. E. & Mulvey, R. E. (2002).J. Organomet. Chem.656, 63–70.
Kristen, M. O., Bildstein, B. & Krajate, A. (2001). Patent [BASF AG] WO 2002064645, EP 0201264, DE 10106902.
Molecular Structure Corporation & Rigaku Corporation (1998).TEXSAN. MSC, The Woodlands, Texas, USA, and Rigaku Corporation, Tokyo, Japan. Zuckerman, R. L., Krska, S. W. & Bergman, R. G. (2000).J. Am. Chem. Soc.
Figure 1
ORTEP-3(Farrugia, 1997) diagram of (I), drawn with 50% probability
supporting information
Acta Cryst. (2006). E62, m950–m951 [https://doi.org/10.1107/S1600536806011007]
Chlorobis(
η
5-cyclopentadienyl)[
N
-(2,6-diisopropylphenyl)-
N
-(1-phenylvinyl)-amide]zirconium(IV)
Benjamin L. Rupert, John Arnold and Alexander Krajete
Chloro[N-(2,6-diisopropylphenyl)-N-(1- phenylvinyl)amide]bis(η5-cyclopentadienyl)zirconium
Crystal data
[ZrCl(C5H5)2(C20H24N)]
Mr = 535.28
Orthorhombic, P212121
Hall symbol: P 2ac 2ab a = 9.5134 (8) Å b = 14.718 (1) Å c = 18.591 (2) Å V = 2603.2 (4) Å3
Z = 4
F(000) = 1112 Dx = 1.366 Mg m−3
Mo Kα radiation, λ = 0.7107 Å Cell parameters from 2777 reflections θ = 2.4–22.6°
µ = 0.54 mm−1
T = 161 K Column, yellow 0.20 × 0.10 × 0.08 mm
Data collection Bruker SMART 1000
diffractometer ω scans
Absorption correction: multi-scan (Blessing, 1995)
Tmin = 0.870, Tmax = 0.958
11464 measured reflections
4287 independent reflections 3249 reflections with I > 3σ(I) Rint = 0.055
θmax = 24.7°
h = 0→11 k = 0→17 l = −21→21
Refinement Refinement on F R[F2 > 2σ(F2)] = 0.039
wR(F2) = 0.039
S = 1.09 3249 reflections 298 parameters
H-atom parameters constrained
w = 1/[σ2(F
o) + 0.00022|Fo|2]
(Δ/σ)max = 0.001
Δρmax = 0.46 e Å−3
Δρmin = −0.68 e Å−3
Absolute structure: Flack (1983), 1807 Friedel pairs
Absolute structure parameter: 0.06 (5)
Special details
Refinement. Refinement using reflections with F2 > 3.0 σ(F2). The weighted R-factor (wR), goodness of fit (S) and R
-factor (gt) are based on F, with F set to zero for negative F.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
H18 0.2357 0.2272 0.1635 0.0321* H19 0.1433 0.1690 0.0577 0.0408* H20 0.0764 0.2647 −0.0345 0.0361* H21 0.2706 0.4598 0.2077 0.0301* H22 0.4615 0.3930 0.2656 0.0394* H23 0.4872 0.3945 0.1832 0.0394* H24 0.4401 0.3053 0.2207 0.0394* H25 0.1905 0.2962 0.2731 0.0384* H26 0.0925 0.3799 0.2657 0.0384* H27 0.2231 0.3845 0.3155 0.0384* H28 0.1314 0.5008 −0.0420 0.0335* H29 0.2224 0.3983 −0.1244 0.0532* H30 0.0944 0.4484 −0.1576 0.0532* H31 0.0776 0.3501 −0.1288 0.0532* H32 −0.1227 0.4067 −0.0527 0.0485* H33 −0.0975 0.5037 −0.0826 0.0485* H34 −0.0985 0.4871 −0.0002 0.0485*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
C25 0.027 (3) 0.023 (3) 0.026 (3) 0.008 (3) −0.006 (2) 0.006 (3) C26 0.026 (3) 0.044 (4) 0.029 (4) −0.003 (3) −0.005 (3) 0.017 (3) C27 0.023 (3) 0.046 (4) 0.027 (4) 0.008 (3) −0.003 (3) 0.012 (3) C28 0.029 (4) 0.031 (3) 0.021 (4) −0.006 (3) −0.005 (2) −0.003 (2) C29 0.060 (4) 0.051 (4) 0.021 (3) −0.008 (5) −0.004 (3) −0.011 (3) C30 0.038 (4) 0.053 (4) 0.032 (4) −0.004 (3) −0.017 (3) 0.014 (3)
Geometric parameters (Å, º)
C8—C9—C10 107.6 (5) H29—C29—H31 109.366 Zr1—C10—C6 74.1 (3) H30—C29—H31 109.492 Zr1—C10—C9 73.7 (3) C28—C30—H32 109.564 C6—C10—C9 108.0 (5) C28—C30—H33 109.678 N1—C11—C12 120.2 (5) C28—C30—H34 109.498 N1—C11—C13 122.8 (5) H32—C30—H33 109.376 C12—C11—C13 117.0 (5) H32—C30—H34 109.292 C11—C13—C14 123.8 (5) H33—C30—H34 109.418 C11—C13—C18 118.8 (5)