Acta Crystallographica Section E
Structure Reports Online
ISSN 1600-5368
endo
-(1
000R
,2
000R
,5
000S
,7
000R
,9
000S
)-2-(9
000-Benzyloxy-2
000-phenyl-3
000,6
000-dioxabicyclo[3.2.2]nonan-7
000-yl)-7-bromo-5,8-di-methoxynaphthalen-1-yl acetate
Timothy J. Brenstrum,
Margaret A. Brimble and Peter Turner*
School of Chemistry, University of Sydney, Camperdown, NSW 2006, Australia.
Correspondence e-mail: p.turner@chem.usyd.edu.au
Key indicators
Single-crystal X-ray study T= 295 K
Mean(C±C) = 0.009 AÊ Rfactor = 0.036 wRfactor = 0.114 Data-to-parameter ratio = 7.8
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2001 International Union of Crystallography
The title compound, (1), is the product of a model synthesis of an analogue of the antibiotic medermycin, and is thought to be the result of an unusual 1,6-hydride shift.
Comment
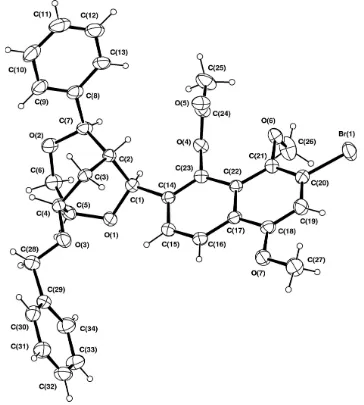
Recent synthetic effort has been directed towards the synth-esis of the pyranonaphthoquinone antibiotic medermycin (2) which was isolated from Streptomyces tanashiensis and contains a C-glycoside linkage to a 2-deoxy sugar (Takanoet al., 1976). As part of this programme we embarked on model studies directed towards the synthesis of the 2-deoxyglucosyl analogue of medermycin, (3). The key step in the approach to (3) involved the direct C-glycosylation of 3-bromonaphthol, (4), with a 2-deoxyglucosyl donor, (5) (Brimble & Brenstrum, 2000) (see Scheme) which was expected to afford the desired C-glycoside, (6). Unfortunately this critical C-glycosylation reaction afforded predominantly C-glycoside (1) wherein extensive rearrangement of the 2-deoxyglucosyl moiety had taken place. The structure of this rearranged C-glycoside was established by X-ray crystallography of the acetate derivative of the initial glycosylation product (Fig. 1).
The structure of this rearranged C-glycoside (1) clearly shows that extensive rearrangement of the carbohydrate skeleton has taken place. This rearrangement has been proposed to occurviaan unusual 1,6-hydride shift similar to that observed by Steel et al. in the dimerization of tri-O -benzyl-d-glucal (Byerleyet al., 1998). The X-ray structure also clearly establishes that the naphthalene ring is endo to the bicylic ring system
Experimental
Trimethylsilyl tri¯uoromethanesulfonate (51 ml, 0.266 mmol) and silver perchlorate (2 mg, 5 mol%) were added to a stirred solution of 3-bromo-1,4-dimethoxy-5-hydroxynaphthalene, (4) (50 mg,
0.177 mmol), and tri-O-benzyl-2-deoxy-d-glucosyl acetate, (5)
(101 mg, 210 mmol), in dry acetonitrile (5 ml) at 273 K. The mixture was stirred for 1 h then quenched with aqueous bicarbonate solution (5 ml). The reaction mixture was extracted with dichloromethane (3
50 ml), washed with water (100 ml) and dried (magnesium sulfate).
The solvent was removed at reduced pressure and the oily residue puri®ed by ¯ash chromatography using hexane-ethyl acetate (4:1) as
eluent to give a mixture of a rearranged C-glycoside andb
-C-glyco-side (6) (71 mg, 6:1). This mixture of glyco-C-glyco-sides was subjected to HPLC.
Triethylamine (0.50 ml, 3.59 mmol), acetic anhydride (0.25 ml, 2.65 mmol) and a catalytic quantity of dimethylaminopyridine were added to a solution of the above rearranged C-glycoside (98 mg, 0.166 mmol) in dichloromethane (2 ml). The solution was stirred overnight then the solvent removed at reduced pressure. The residue was puri®ed by ¯ash chromatography using hexane-ethyl acetate (4:1) to give the acetate (1) (99 mg, 94%) which was recrystallized from hexane-ethyl acetate to give pale-brown needles (m.p. 477± 478 K).
Crystal data
C34H33BrO7
Mr= 633.51
Orthorhombic,P212121
a= 19.5150 (10) AÊ
b= 23.667 (2) AÊ
c= 6.541 (2) AÊ
V= 3021.0 (10) AÊ3
Z= 4
Dx= 1.393 Mg mÿ3
CuKradiation Cell parameters from 25
re¯ections
= 26.6±30.2 = 2.25 mmÿ1
T= 294 (2) K Acicular, pale brown 0.550.130.08 mm
Data collection
Rigaku AFC-7Rdiffractometer
!±2scans
Absorption correction: scan (Northet al., 1968)
Tmin= 0.715,Tmax= 0.835
2962 measured re¯ections 2962 independent re¯ections 2068 re¯ections withI> 2(I)
max= 65.0
h= 0!22
k= 0!27
l= 0!7
3 standard re¯ections every 150 re¯ections intensity decay: 2.4%
Re®nement
Re®nement onF2
R[F2> 2(F2)] = 0.036
wR(F2) = 0.114
S= 1.02 2962 re¯ections 382 parameters
H-atom parameters constrained
w= 1/[2(F
o2) + (0.0579P)2
+ 0.6329P]
whereP= (Fo2+ 2Fc2)/3
(/)max< 0.001
max= 0.31 e AÊÿ3
min=ÿ0.34 e AÊÿ3
Absolute structure: Flack (1983) and Bernardinelli & Flack(1985); no Friedel pairs
Flack parameter =ÿ0.06 (3)
Data collection and cell re®nement: MSC/AFC Diffractometer
Control Software (Molecular Structure Corporation, 1995); data
reduction: TEXSAN (Molecular Structure Corporation, 1992);
structure solution: SIR92 (Altomare et al., 1993); structure
re®ne-ment:SHELXL97 (Sheldrick, 1997); molecular graphics:TEXSAN
(Molecular Structure Corporation, 1997).
References
Altomare, A., Cascarano, M., Giacovazzo, C. & Guagliardi, A. (1993).J. Appl. Cryst.26, 343.
Bernardinelli, G. & Flack, H. D. (1985).Acta Cryst.A41, 500±511. Brimble, M. A. & Brenstrum, T. J. (2000)Tetrahedron Lett.pp. 1107±1110. Byerley, A. L. J., Kenwright, A. M., Lehman, C. W., MacBride, J. A. H. & Steel,
P. G. (1998).J. Org. Chem.63, 193±194. Flack, H. D. (1983).Acta Cryst.A39, 876±881.
Hall, S. R., du Boulay, D. J. & Olthof-Hazekamp, R. (1999). Editors.Xtal3.6 System. University of Western Australia, Australia.
Johnson, C. K. (1976).ORTEPII. Report ORNL-5138. Oak Ridge National Laboratory, Tennessee, USA.
Molecular Structure Corporation (1992).TEXSAN. UNIX Version 1.8. MSC, 3200 Research Forest Drive, The Woodlands, TX 77381, USA.
Molecular Structure Corporation (1995).MSC/AFC Diffractometer Control Software. MSC, 3200 Research Forest Drive, The Woodlands, TX 77381, USA.
Molecular Structure Corporation (1997).TEXSAN for Windows.MSC, 3200 Research Forest Drive, The Woodlands, TX 77381, USA.
North, A. C. T., Phillips, D. C. & Mathews, F. S. (1968).Acta Cryst.A23, 351± 359.
Sheldrick, G. M. (1997).SHELXL97. University of GoÈttingen, Germany. Takano, S., Hasuda, K., Ito, A., Koide, Y., Ishii, F., Haneda, I., Chihara, S. &
Koyami, T. (1976).J. Antibiot.28, 765±768.
Acta Cryst.(2001). E57, o28±o29 Timothy J. Brenstrumet al. C34H33BrO7
o29
organic papers
Figure 1
supporting information
Acta Cryst. (2001). E57, o28–o29 [doi:10.1107/S1600536800018092]
endo
-(1
′
R
,2
′
R
,5
′
S
,7
′
R
,9
′
S
)-2-(9
′
-Benzyloxy-2
′
-phenyl-3
′
,6
′
-dioxabicyclo-[3.2.2]nonan-7
′
-yl)-7-bromo-5,8-dimethoxynaphthalen-1-yl acetate
Timothy J. Brenstrum, Margaret A. Brimble and Peter Turner
S1. Comment
Recent synthetic effort has been directed towards the synthesis of the pyranonaphthoquinone antibiotic medermycin (2)
which was isolated from Streptomyces tanashiensis and contains a C-glycoside linkage to a 2-deoxy sugar (Takano et al.,
1976). As part of this programme we embarked on model studies directed towards the synthesis of the 2-deoxyglucosyl
analogue of medermycin (3). The key step in the approach to (3) involved the direct C-glycosylation of 3-bromonaphthol
(4) with a 2-deoxyglucosyl donor (5) (Brimble and Brenstrum, 2000) (Scheme 1) which was expected to afford the
desired C-glycoside (6). Unfortunately this critical C-glycosylation reaction afforded predominantly C-glycoside (1)
wherein extensive rearrangement of the 2-deoxyglucosyl moiety had taken place. The structure of this rearranged
C-glycoside was established by X-ray crystallography of the acetate derivative of the initial glycosylation product (Fig. 1).
The structure of this rearranged C-glycoside (1) clearly shows that extensive rearrangement of the carbohydrate
skeleton has taken place. This rearrangement has been proposed to occur via an unusual 1,6-hydride shift similar to that
observed by Steel et al. in the dimerization of tri-O-benzyl-D-glucal (Byerley et al.,1998). The X-ray structure also
clearly establishes that the naphthalene ring is endo to the bicylic ring system
S2. Experimental
Trimethylsilyl trifluoromethanesulfonate (51 ml, 0.266 mmol) and silver perchlorate (2 mg, 5 mol%) were added to a
stirred solution of 3-bromo-1,4-dimethoxy-5-hydroxynaphthalene (4) (50 mg, 0.177 mmol) and tri-O-benzyl-2-deoxy-D
-glucosyl acetate (5) (101 mg, 210 mmol) in dry acetonitrile (5 ml) at 0 °C. The mixture was stirred for 1 h then quenched
with aqueous bicarbonate solution (5 ml). The reaction mixture was extracted with dichloromethane (3 x 50 ml), washed
with water (100 ml) and dried (magnesium sulfate). The solvent was removed at reduced pressure and the oily residue
purified by flash chromatography using hexane-ethyl acetate (4:1) as eluent to give a mixture of a rearranged C-glycoside
and b-C-glycoside (6) (71 mg, 6:1). This mixture of glycosides was subjected to HPLC.
Triethylamine (0.50 ml, 3.59 mmol), acetic anhydride (0.25 ml, 2.65 mmol) and a catalytic quantity of
dimethylamino-pyridine were added to a solution of the above rearranged C-glycoside (98 mg, 0.166 mmol) in dichloromethane (2 ml).
The solution was stirred overnight then the solvent removed at reduced pressure. The residue was purified by flash
chromatography using hexane-ethyl acetate (4:1) to give the acetate (1) (99 mg, 94%) which was recrystallized from
supporting information
sup-2
[image:4.610.124.488.72.477.2]Acta Cryst. (2001). E57, o28–o29
Figure 1
ORTEP (Johnson, 1976, Hall et al. 1999) projection of (1) with displacement ellipsoids shown at the 20% level.
(1)
Crystal data C34H33BrO7 Mr = 633.51
Orthorhombic, P212121 a = 19.515 (1) Å b = 23.667 (2) Å c = 6.541 (2) Å V = 3021.0 (10) Å3 Z = 4
F(000) = 1312
Dx = 1.393 Mg m−3
Cu Kα radiation, λ = 1.54178 Å Cell parameters from 25 reflections θ = 26.6–30.2°
µ = 2.25 mm−1 T = 294 K
Data collection Rigaku AFC7R
diffractometer
Radiation source: rotating anode Graphite monochromator ω–2θ scans
Absorption correction: ψ scan (North et al., 1968)
Tmin = 0.715, Tmax = 0.835 2962 measured reflections
2962 independent reflections 2068 reflections with I > 2σ(I) Rint = 0.000
θmax = 65.0°, θmin = 2.9° h = 0→22
k = 0→27 l = 0→7
3 standard reflections every 150 reflections intensity decay: 2.4%
Refinement Refinement on F2 Least-squares matrix: full R[F2 > 2σ(F2)] = 0.036 wR(F2) = 0.114 S = 1.02 2962 reflections 382 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H-atom parameters constrained w = 1/[σ2(F
o2) + (0.0579P)2 + 0.6329P] where P = (Fo2 + 2Fc2)/3
(Δ/σ)max < 0.001 Δρmax = 0.31 e Å−3 Δρmin = −0.34 e Å−3
Absolute structure: Flack (1983) and
Bernardinelli & Flack(1985); No Friedel pairs Absolute structure parameter: −0.06 (3)
Special details
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
supporting information
sup-4
Acta Cryst. (2001). E57, o28–o29
H28A 0.4470 0.1951 0.8947 0.102* H28B 0.4875 0.1986 1.1022 0.102* C29 0.4722 (3) 0.1169 (2) 1.0074 (9) 0.0675 (14) C30 0.4775 (3) 0.0943 (3) 1.2008 (10) 0.0789 (16) H30 0.4909 0.1173 1.3092 0.095* C31 0.4632 (4) 0.0379 (3) 1.2355 (12) 0.0911 (19) H31 0.4657 0.0231 1.3670 0.109* C32 0.4453 (3) 0.0042 (3) 1.0755 (15) 0.094 (2) H32 0.4352 −0.0336 1.0984 0.113* C33 0.4421 (4) 0.0251 (3) 0.8851 (14) 0.097 (2) H33 0.4302 0.0014 0.7772 0.116* C34 0.4565 (4) 0.0818 (3) 0.8475 (11) 0.0880 (19) H34 0.4553 0.0958 0.7147 0.106*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
supporting information
sup-6
Acta Cryst. (2001). E57, o28–o29
C24 0.056 (3) 0.062 (3) 0.084 (4) 0.002 (2) 0.005 (3) 0.021 (3) C25 0.082 (4) 0.087 (4) 0.143 (7) −0.001 (3) 0.016 (5) 0.061 (5) C26 0.092 (5) 0.165 (7) 0.052 (3) 0.030 (5) −0.005 (3) 0.013 (4) C27 0.099 (4) 0.073 (4) 0.117 (6) 0.019 (3) 0.014 (4) 0.033 (4) C28 0.095 (4) 0.069 (3) 0.091 (4) −0.013 (3) 0.024 (4) 0.002 (3) C29 0.069 (3) 0.062 (3) 0.072 (4) −0.011 (2) 0.015 (3) −0.005 (3) C30 0.090 (4) 0.071 (3) 0.076 (4) 0.001 (3) 0.013 (3) −0.005 (3) C31 0.107 (5) 0.071 (4) 0.095 (5) −0.002 (4) 0.016 (5) 0.010 (4) C32 0.084 (4) 0.062 (3) 0.135 (7) −0.012 (3) 0.013 (5) 0.013 (5) C33 0.100 (5) 0.074 (4) 0.117 (6) −0.020 (3) −0.004 (4) −0.019 (4) C34 0.109 (5) 0.080 (4) 0.075 (4) −0.016 (4) −0.004 (4) −0.002 (3)
Geometric parameters (Å, º)
C9—H9 0.9300 C31—H31 0.9300 C10—C11 1.381 (10) C32—C33 1.341 (11) C10—H10 0.9300 C32—H32 0.9300 C11—C12 1.369 (12) C33—C34 1.393 (9) C11—H11 0.9300 C33—H33 0.9300 C12—C13 1.365 (10) C34—H34 0.9300
supporting information
sup-8
Acta Cryst. (2001). E57, o28–o29
H6A—C6—H6B 107.7 O7—C27—H27B 109.5 O2—C7—C8 108.0 (5) H27A—C27—H27B 109.5 O2—C7—C2 112.3 (4) O7—C27—H27C 109.5 C8—C7—C2 111.2 (4) H27A—C27—H27C 109.5 O2—C7—H7 108.4 H27B—C27—H27C 109.5 C8—C7—H7 108.4 O3—C28—C29 108.9 (5) C2—C7—H7 108.4 O3—C28—H28A 109.9 C13—C8—C9 117.2 (6) C29—C28—H28A 109.9 C13—C8—C7 121.0 (6) O3—C28—H28B 109.9 C9—C8—C7 121.7 (5) C29—C28—H28B 109.9 C8—C9—C10 120.8 (7) H28A—C28—H28B 108.3 C8—C9—H9 119.6 C34—C29—C30 118.9 (5) C10—C9—H9 119.6 C34—C29—C28 120.7 (6) C11—C10—C9 119.8 (8) C30—C29—C28 120.4 (6) C11—C10—H10 120.1 C29—C30—C31 120.6 (7) C9—C10—H10 120.1 C29—C30—H30 119.7 C12—C11—C10 119.4 (8) C31—C30—H30 119.7 C12—C11—H11 120.3 C32—C31—C30 119.4 (7) C10—C11—H11 120.3 C32—C31—H31 120.3 C13—C12—C11 120.4 (7) C30—C31—H31 120.3 C13—C12—H12 119.8 C33—C32—C31 120.7 (6) C11—C12—H12 119.8 C33—C32—H32 119.6 C12—C13—C8 122.3 (8) C31—C32—H32 119.6 C12—C13—H13 118.8 C32—C33—C34 120.6 (7) C8—C13—H13 118.8 C32—C33—H33 119.7 C23—C14—C15 118.7 (4) C34—C33—H33 119.7 C23—C14—C1 119.7 (4) C29—C34—C33 119.6 (7) C15—C14—C1 121.5 (4) C29—C34—H34 120.2 C16—C15—C14 121.1 (5) C33—C34—H34 120.2 C16—C15—H15 119.5