Gene structure, phytogeny and mutation analysis
of RING3 - a novel MHC-encoded gene
A th e sis p re se n te d fo r th e d e g re e o f D o c to r o f P h ilo so p h y b y
Karen Louise Thorpe
University of London
Im p e ria l C a n c e r R e se a rc h F u n d
4 4 , L in c o ln ’s In n F ield s
ProQuest Number: 10015797
All rights reserved
INFORMATION TO ALL USERS
The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. Also, if material had to be removed,
a note will indicate the deletion.
uest.
ProQuest 10015797
Published by ProQuest LLC(2016). Copyright of the Dissertation is held by the Author.
All rights reserved.
This work is protected against unauthorized copying under Title 17, United States Code. Microform Edition © ProQuest LLC.
ProQuest LLC
789 East Eisenhower Parkway P.O. Box 1346
ABSTRACT
The hum an major histocom patibility com plex (MHC), on the short arm of
chrom osom e 6, contains a cluster of genes w hich show diverse immune
functions, like the recognition of foreign antigen and the generation o f an
inflam m atory response. However, the RINGS gene shows no obvious immune
function despite its location in the centre of the MHC. RINGS was found to
span 12 kb and contains IS exons, with two alternative start codons. The
RINGS gene was also isolated in the m ouse and m apped to a syntenic position
in the murine MHC. Com parison of the RINGS gene structure in human,
m ouse and chicken revealed a perfect conservation in the intron/exon
boundaries o f the coding exons and high conservation at the am ino acid level.
The ORFX gene, a hum an hom ologue o f RINGS, was isolated and m apped to
9qS4. This gene shows a highly conserved gene structure but is almost three
times larger than RINGS, mainly due to the presence o f repeat elements.
Evolutionary analysis indicates that RINGS may be over 1 bilhon years old
w ith hom ologues isolated in yeast, worm, fly, fish, chicken, m ouse and hum an.
The gene structure has been conserved for at least S50 million years an d
linkage to the M HC has been established in five vertebrate species. E vidence
also suggests that ORFX and RINGS arose by an ancient duplication S 5 0 -
400 m illion years ago. Both RINGS and ORFX are ubiquitously expressed in
hum an adult and foetal tissues, with a highly abundant expression in th e
testis. Human and m ouse have three variant RINGS transcripts, one o f w hich
is purely testis-specific. Growing evidence suggests that RINGS may be a
nuclear kinase w ith a raised activity in certain types o f leukaemia. M u tatio n
analysis o f the RINGS coding regions in ALL, CM L and normal individuals,
revealed a low level of polymorphism across the gene. However, for o n e
deletion mutation, a significant deviation from H ardy-W einberg proportions
ACKNOWLEDGMENTS
" I t was tfie 6est o f times, it was tRe worst o f times.
I t was tRe age o f wisdom, it was tRe age offooCisRness”
A Tale of Two Cities, Charles Dickens, 1859.
The above quotation is from one of my favourite pieces of literature and the words encapsulate the very essence of my Ph.D. research. I have been extremely fortunate to have worked in two of the finest research institutes during the course of my Ph D: the Imperial Cancer Research Fund and the Sanger Centre. Without their funding this research would not have been possible.
I wish to thank my supervisors: Stephan Beck for his tireless enthusiasm, encouragement and support of my project and Jonathan Wolfe for helpful suggestions and advice. A big thank you to all members of the chromosome 6 sequencing team at the Sanger Centre; I am grateful for your help and encouragement. Many thanks to Peter Rice (Sanger Centre) and Peter Woollard (HGMP) for their invaluable assistance with the phylogenetic analysis. A special thank you is also extended to Patricia Gorman, Jill Williamson and Denise Sheer of the Human Cytogenetics Laboratory, ICRF, London, for their help with the FISH analysis. Many thanks to past colleagues at ICRF: Ivo Gut, Liz Radley, William Newell, Claire Thomas and Louise Hosking. I also wish to thank members of the Immunogenetics lab at Cambridge University: John Trowsdale, Derek McCusker and Ruma Raha-Chowdhury. My gratitude is extended to Alan Schafer and members of the mutation analysis group at Hexagen, Cambridge for allowing me to use their facilities and learn the art of DNA mutation detection. Many thanks are extended to John Trowsdale, Mary Carrington and Tevfik Dorak; it has been a pleasure to work with you all.
I dedicate this thesis to my parents, Sandra and Richard, who encouraged me to continue when the going was tough and were proud of my smallest and largest achievements. I also dedicate this to Keir who has changed my life and who understands the highs and lows of being a Ph.D. student.
TABLE OF CONTENTS
Title page.
Abstract...
Acknowledgments.
Chapter 1
Introduction: The human major histocompatibility complex
and introducing the “Really Interesting New Gene 3”
(RING3)... 14
The human major histocompatibility complex... 14
History of the human M HC...15
Physical mapping of the M HC... 16
Genes of the human M HC... 18
Class I genes ...18
Class HI g en es... 18
Class II g en es...19
Structure of class I and class II MHC molecules... 20
Class I stru ctu re...21
Class II stru ctu re... 21
MHC class I and class II gene structure...22
Antigen presentation by class I and class II molecules... 22
Class I antigen presenation... 22
Class II antigen presenation... 23
Comparative genomics and evolution of the MHC... 24
Sequencing of the human major histocompatibility complex... 27
Mapping and cloning of the human RINGS cDNA... 28
Sequencing of the RINGS cDNA...29
S o lu tio n s ... 32
M e th o d s... 40
Production of M13 shotgun library...40
Isolation o f fragm ents fo r subcloning... 41
Fragment s e lf ligation...42
S o n ic a tio n...43
Sub-fragment end repair... 44
Size fra c tio n a tio n... 44
Cloning into M13 vectors... 46
Preparation o f Sma I -restricted M lS m p lS... 47
L i g a t io n...48
Preparation o f competent cells...49
T r a n s fo r m a tio n...50
Production of M l3 phage master plate... 52
Thermoextraction of M l3 template DNA... 52
Triton preparation of M l3 template DNA... 54
Autom ated DNA sequencing...56
D y e - p r im e r s... 57
D y e - te r m in a to r s... 57
In s tr u m e n ta tio n...58
Instrumentation o f the ABI prism ™ 373A and 377 automated DNA seq u en cers...58
DNA cycle sequencing with fluorescent dye primers (without the use o f ready-mix kits)...61
Dye terminator cycle sequencing using the ABI PRISM ™ ready reaction kit (AmpHTaq DNA polymerase FS)... 63
Shotgun sequencing with dye terminators... 63
Sequencing PGR products with dye terminators... 64
DNA sequencing with Amersham DYEnamic ET dye p r i m e r s... 64
DNA sequence projects - analysis and contiguation...65
DNA sequence analysis... 65
Finishing o f a DNA sequencing project...66
Labelling DNA fragments with [a^^P]-dCTP by the random prim er extension m ethod... 67
Purification o f radiolabelled DNA probe through a Sephadex colum n... 68
Preparation o f plating bacteria... 70
Plating the bacteriophage X129/svJ library... 70
Plaque lifting onto nylon filte rs... 72
Hybridisation o f the X129/svJ plaque lift filters...73
Identification o f positively hybridising plaques... 75
Picking positive plaques... 76
Secondary and tertiary screening o f the X129/svJ library...76
Preparation o f X DNA from plate lysates...77
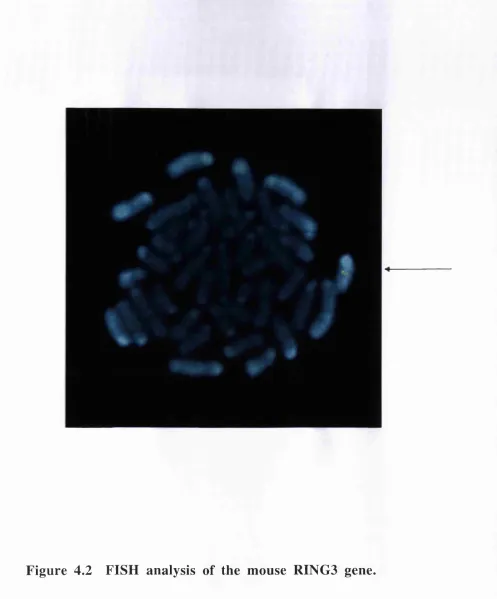
DNA sequencing and analysis o f mouse RINGS clone IIK L T . 78 Fluorescent in situ hybridisation (FISH) mapping of the mouse RINGS clone I I K L T ... 78
Transcription pattern of mouse and human RINGS genes and the ORFX g e n e ...79
Generation and labelling o f probes... 79
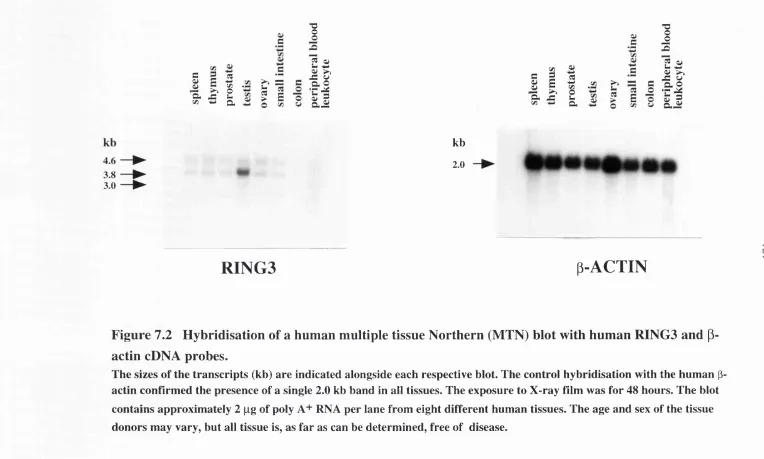
Hybridisation o f human and mouse multiple tissue Northern b l o t s...80
Generation o f ORFX and RINGS specific probes...81
Hybridisation o f RNA blot with ORFX and RINGS specific p r o b e s...82
Generation and labelling o f probes fo r the ORFX MTN screen 8S Cellular localisation of the RINGS and ORFX proteins... 8S Design o f p ep tides...8S Gluteraldehyde coupling o f peptides... 84
Immunisation o f coupled peptides... 85
Whole cell enzyme-linked immunosorbant assay (ELISA)... 85
SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and W estern b lo ttin g... 86
Protein minigel fo r western blotting...87
Dry blotting o f SDS-protein gels... 88
Probing o f Western blot membranes... 89
Cellular localisation o f RINGS and ORFX by immuno flu o rescen ce sta in ing...90
Viewing the slides by fluorescent microscopy... 91
Cloning, mapping and sequencing of the ORFX gene... 91
Isolation o f ORFX positive clone H 2AIK LT... 91
Fluorescent in situ hybridisation (FISH) mapping o f the human ORFX gene... 92
Microsatellite typing of the (GT)n repeat in the human RING3
g e n e ... 94
DNA sa m p les...94
P rim er d esig n...94
End-labelling o f the 5 ’-primer with [y- ^^PJATP...94
PCR amplification and electrophoresis o f the microsatellite r e p e a t...94
Statistical analysis o f the ( GT)n micro satellite...95
Mutation analysis of the RING3 gene by high-throughput flu o rescen t SSC P ... 96
DNA samples and isolation...96
Primer design and PCR optimisation/amplification... 96
Gel electrophoresis...97
Data a n a lysis... 98
S tatistical analysis... 98
DNA sequence analysis...98
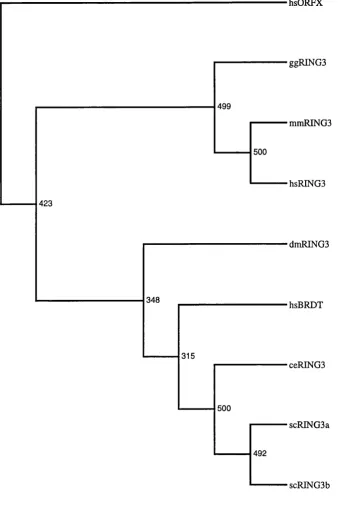
Multiple sequence alignment and phylogenetic analysis... 99
M ultiple sequence alignm ent...99
Phylogenetic analysis using a protein distance method...99
Phylogenetic analysis using a maximum likelihood method... 101
Chapter 3
Large-scale sequencing of the MHC class II region:
mapping, sequencing and analysis of the human RINGS
gene... 103
In tro d u c tio n ...103
R e s u lts ... 105
D is c u ss io n ... 106
Chapter 4
Cloning, mapping and sequencing of the mouse RING3
gene... 113
In tro d u c tio n ...113
R e s u lts ... 115
Isolation and mapping of the mouse RING3 gene, mmRING3...115
R epeat an a ly sis... 117
D is c u ss io n ... 117
Chapter 5
Cloning, mapping and sequencing of the human ORFX
gene... 125
In tro d u c tio n ... 125
R e s u lts ...126
Chrom osom al lo calisatio n ...126
G ene stru c tu re ... 126
R epeat an aly sis... 127
D is c u ss io n ...128
Chapter 6
Comparative genomics and evolutionary analysis of
RING3... 136
In tro d u c tio n ... 136
R e s u lts ...138.
Gene structure of RING3 homologues...138
Sequence identity and multiple sequence alignment... 139
Phylogenetic analysis of RING3 homologues...140
D is c u ss io n ...143
Chapter 7
Transcription pattern of the human and mouse RING3
genes and the ORFX gene: identification of variant RING3
transcripts. Cellular localisation of the human RING3 and
ORFX gene products...
161
In tro d u c tio n ... 161
R e s u lts ...163
Transcription pattern of the human and mouse RING3 genes and the ORFX g e n e ...163
Chapter 8
Microsatellite typing of the (GT)n repeat in the human
RING3gene... 182
In tro d u c tio n ... 182
R e s u lts ... 184
Microsatellite typing of the (GT)„ dinucleotide repeat... 184
D is c u ss io n ... 185
Chapter 9
Detection of polymorphism in the RING3 gene by
high-throughput fluorescent SSCP analysis...193
In tro d u c tio n ... 193
R e s u lts ... 195
High-throughput mutation detection... 195
Polymorphism and disease association... 196
D is c u ss io n ... 198
Chapter 10
Concluding remarks... 210
S u m m ary ... 210
Future co n sid eratio n s... 210
Appendix... 213
EMBL accession numbers for submitted sequences...213
EMBL accession numbers for sequences cited in the text as containing brom odom ains...213
TABLES AND FIGURES
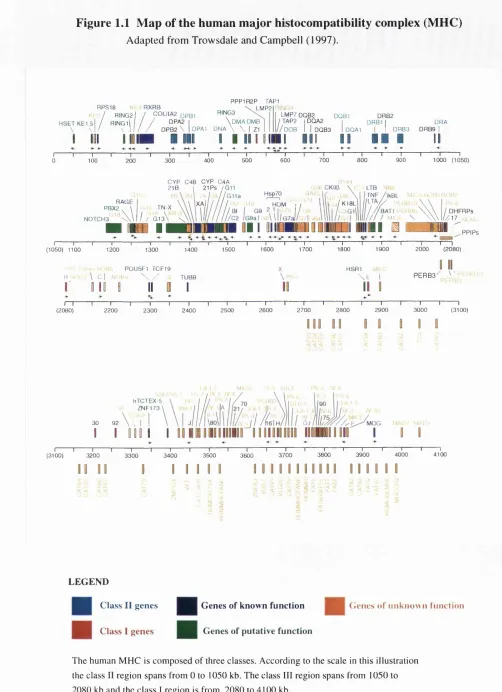
Figure 1.1 Map of the human major histocompatibility complex... 31
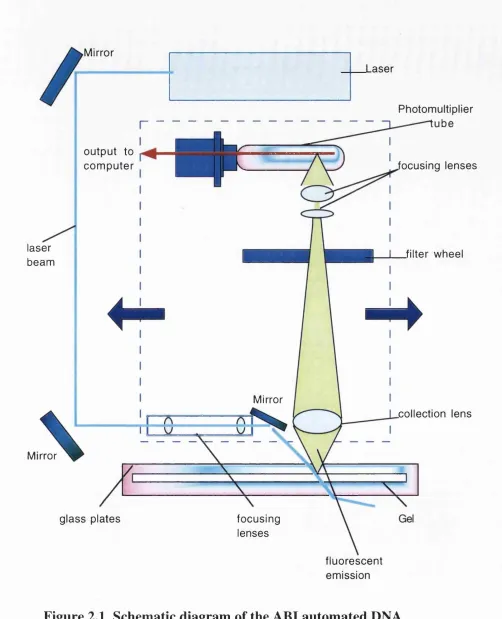
Figure 2.1 Schematic diagram of the ABI automated DNA sequencer... 59
Figure 3.1 Nucleotide and deduced amino acid sequence of the RING3 cDNA clone C E M 32... 109
Figure 3.2 Clone map of the RING3 locus... 110
Figure 3.3 Dot matrix plot of human RING3 cDNA sequence versus human RING3 genom ic sequence... I l l Figure 3.4 Gene structure of the human RING3 gene (hsRING3)... 112
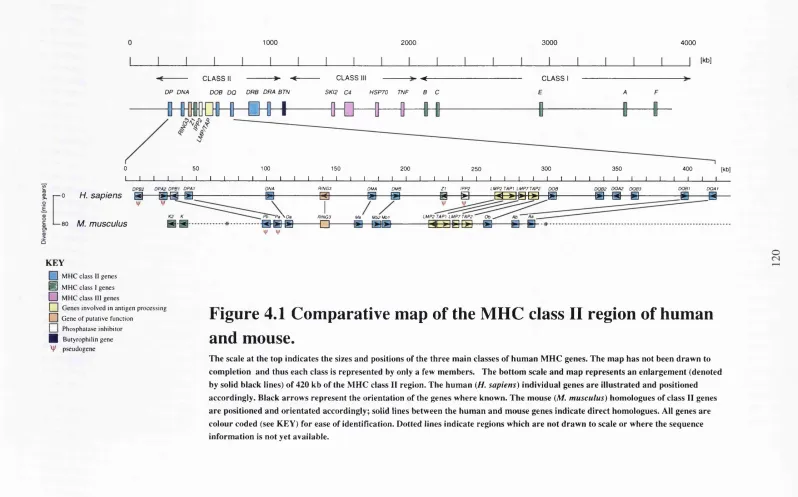
Figure 4.1 Comparative map of the MHC class II region of human and mouse 120 Figure 4.2 FISH analysis of the mouse RING3 gene... 121
Figure 4.3 Gene structures of RING3 homologues... 122
Figure 4.4 Amino acid alignment of RING3 homologues... 123
Table 4.1 Amino acid changes between the human and mouse RING3 sequences... 124
Figure 5.1 FISH analysis of the human ORFX gene... 132
Figure 5.2 Comparison of the gene structures of ORFX and RENG3...133
Figure 5.3 Predicted transcription factor binding sites for NFkB, Bicoid (Bed) and Kriippel (Kr) in the genomic sequences of hsORFX, hsRING3 and mmRING3... 134
Figure 6.1 Gene structures of RINGS homologues... 150
Table 6.1 Amino acid identities (given as a percentage) between RINGS
h o m o lo g u e s... 151
Figure 6.2 Amino acid sequence alignment of RINGS homologues... 152
Figure 6.S Amino acid sequence alignment of the S’-termini of the hsbrdt and
dmRINGS g e n e s...155
Figure 6.4 Multiple sequence alignment of the bromodomain...156
Figure 6.5 Consensus cladogram (unrooted) derived from the phylogenetic analysis
of RINGS hom ologues... 157
Figure 6.6 Consensus cladogram (unrooted) derived from the phylogenetic analysis
of RINGS homologues deleted for the bromodomains...158
Figure 6.7 Phylogenetic tree (unrooted) of RINGS homologues...159
Figure 6.8 Phylogenetic tree (unrooted) of RINGS homologues deleted for the
b ro m o d o m a in s... 160
Figure 7.1 Hybridisation of a human RNA Master blot with human (a) RINGS,
(b) ORFX and (c) ubiquitin gene-specific probes...170
Figure 7.2 Hybridisation of a human multiple tissue Northern (MTN) blot with
human RINGS and p-actin cDNA probes...171
Figure 7.S Hybridisation of a human muliple tissue (MTN) Northern blot with
human RINGS and p-actin cDNA probes...172
Figure 7.4 Hybridisation of a human cancer cell line Northern blot with human
RINGS and P-actin cDNA probes...17S
Figure 7.5 Hybridisation of a mouse multiple tissue Northern (MTN) blot with
Figure 7.6 Hybridisation of a mouse embryo multiple tissue Northern (MTN)
blot with mouse RINGS and human p-actin cDNA probes... 175
Figure 7.7 Hybridisation of human multiple tissue and cancer cell line Northern
(MTN) blots with a human ORFX-specific probe... 176
Figure 7.8 Dot matrix plot of human RINGS cDNA sequence versus human
RINGS genom ic sequence... 177
Figure 7.9 Western blot analysis of the RINGS gene product... 178
Figure 7.10 Western blot analysis of the ORFX gene product... 179
Figure 7.11 Cellular localisation of the RINGS gene product by immuno-
flu o rescen t sta in in g ... 180
Figure 7.12 Cellular localisation of the ORFX gene product by immuno-
flu o rescen t sta in in g ... 181
Figure 8.1 Microsatellite typing of the RINGS (GT)„ repeat in acute lympho
blastic leukaem ia (ALL) DNA samples... 188
Figure 8.2 Microsatellite typing of the RINGS (GT)„ repeat in chronic myeloid
leukaem ia (CML) DNA sam ples...189
Figure 8.S Microsatellite typing of the RINGS (GT)„ repeat in normal (control)
D N A sa m p le s... 190
Table 8.1 Genotypes observed for the RINGS (GT)„ microsatellite repeat in acute
lymphoblastic leukaemia (ALL), chronic myeloid leukaemia (CML) and normal
D N A sa m p le s... 191
Table 8.2 Allele frequencies for the RINGS (GT)„ microsatelhte repeat in ALL,
CM L and norm al sam ples...192
Table 9.1 PCR primers and sequences tested in the SSCP screen of RINGS... 202
the respective nucleotide changes... 203
Table 9.3 Genotypes observed for individual samples at the six variant regions of
R IN G 3 ... 204
Table 9.4 Genotypes and frequencies observed in the SSCP screen for the six
v arian t re g io n s ...205
Table 9.5 Summary of the statistical analysis for the variation at the rg3Hsl40
lo c u s ... 206
Table 9.6 Results of the heterogeneity test between Normal vs ALL samples at the
rg3H s280 lo c u s...207
Figure 9.1 SSCP patterns observed for the rg3Hsl40 amplimers and analysed by
the Genotyper™ softw are...208
CHAPTER 1
I
n t r o d u c t i o n:
T
h e h u m a n m a j o r h i s t o c o m p a t i b i l i t y c o m p l e xAND INTRODUCING THE “ REALLY INTERESTING N E W
G
ene
3” (RINGS)
T
h e h u m a n m a jo r h ist o c o m pa t ib il it y c o m pl e xThe human major histocompatibility complex (MHC) is a 4 Mb region located on the
short arm of chromosome 6 (6p21.3). It contains a cluster of genes, some of which are responsible for recognising foreign antigens in the body and ilhciting an adaptive
immune response to fight these antigens. To date, more than 200 genes have been
located to the human MHC and they show a wide variety of immune and non-immune
functions. The MHC was originally discovered in a study on tumour rejection in mice
and the region remains biomedically important because of its extensive contribution to the acceptance and rejection of tissues during transplantation. The MHC also shows a
strong genetic linkage with the susceptibility to several autoimmune disorders such as
systemic lupus erythematosus, multiple sclerosis, rheumatoid arthritis and insulin-
dependent diabetes mellitus (Thomson, 1995; Hall and Bowness, 1996). In some
instances the susceptibility is caused by variation in the classical MHC antigen-
presenting molecules themselves but polymorphism in other MHC-related genes could
also be contributing towards autoimmunity. Genes which are responsible for causing
some of the commonest hereditary disorders are also located in the MHC e.g. steroid
21-hydroxylase and haemochromatosis. A deficiency of the former gene causes an
adrenogenital disorder and the latter is a common autosomal-recessive disease which
causes abberrant iron metabolism (Feder et a l, 1996). The hereditary disorder
narcolepsy also has a clearly established association with certain MHC class 11 alleles
but the gene has yet to be isolated.
The MHC is one of the most gene-dense regions and represents approximately 1/lOOOth
of the human haploid genome. This chapter aims to discuss the history, genomic
organisation, evolution and the function of the human MHC. The genes of the MHC
novel structures and functions. One such novel gene, called RING3, was mapped to the
middle of the human MHC cluster (Beck et a l, 1992). The gene structure, comparative
genomics, evolution and polymorphism of RING3 are the subject of this thesis.
H
ist o r y o f th e h u m a n m h cIn the 1900s, Ernest E. Tyzzer and Clarence C. Little were the first to demonstrate that
the successful growth of a tumour, transplanted from one inbred strain of mouse to a
second independent strain, was dependent upon several genes (approximately 14 or 15
loci) in the host and the donor— i.e. tumour suscepitibility was a polygenic trait. (Little
and Tyzzer, 1916; Klein, 1986). However, the nature of these susceptibihty genes
remained obscure until Peter A. Gorer discovered a correlation between tumour
transplantation and blood group antigens in 1937. The membranes of red blood cells
(erythrocytes) differ between individuals in their biochemical constituents and as such act as antigens. Antigens are readily detected by antibodies in the serum and extensive
experimentation with these antibodies allowed the blood groups to be defined (for
review see Klein, 1986). Gorer transplanted tumours between three different mouse cell
lines and determined the blood types of those that were susceptible. He showed that the
inheritance of a readily detectable blood antigen, called antigen II, could be correlated
with the susceptibility to tumours (Gorer, 1937). An increasing number of blood
antigens were correlated with tumour susceptibility and for a while it was suggested that
the genes controlling these two phenomena were identical. It was George D. Snell who
embarked on the separation of the tumour susceptibility loci (genes) by performing extensive backcrosses in mouse strains differing in their tumour phenotype (Snell,
1948). Snell proposed that the genes involved in the tumour resistance/susceptibility
should be called histocompatibility (H) genes and since one of these genes was clearly
associated with the antigen II locus, the entire locus was designated histocompatibility-2
or H-2 (Gorer et a l, 1948). As more H genes were discovered it transpired that the
different antigens which they controlled could be “weak” or “strong” in the ability to
resist tumour transplants. The terms major (strong) and minor (weak) became
synonymous with describing the H genes and in later years it was discovered that the
major H genes were found in a complex on chromosome 17 of the mouse. Such a
complex was also found in other species and the term major histocompatibility complex
(MHC) was coined. The H-2 complex is thus the mouse MHC and the minor H genes
were in contrast found to be scattered over the murine genome (Klein, 1986).
The discovery of the human equivalent of the murine H-2 complex was preceded by
two observations. Dausset (1958) reported that patients who had undergone multiple
not to others. Secondly, women who had undergone multiple pregnancies produced maternal antibodies which were capable of agglutinating the leukocytes of some of their
babies (Payne and Rolfs, 1958). The leukocyte agglutinins from the multiparous
women were used to establish the first leukocyte antigen system called Group 4 (Van
Rood and Van Leeuwan, 1963). Large family studies soon established that the different
antigens were controlled by a single complex of loci analogous to the mouse H-2
complex. Initially three linked loci were identified and the name human leukocyte
antigen or HLA was adopted to describe the genes of the human MHC. The first three
identified loci were HLA-A, -B and -C and were later found to encode the human
class I MHC antigens. A fourth human HLA locus was discovered when
histocompatibility tests were performed in vitro. When lymphocytes from unrelated
individuals are cultured together in the same tube the cells undergo cell division and
morphological change—the so-called mixed lymphocyte reaction (MLR). MLR was
found to be controlled by genes that were in the HLA complex but were distinct from
the HLA-A, -B and -C genes. The HLA-D was established as the MLR controlling
locus and was also found to be closely linked to the other HLA loci (McDevitt et ah,
1972). The HLA-D locus was later further subdivided into three loci known as HLA-
DP, -DQ and -DR, encoding the MHC class II antigens. The HLA complex was located
on the short arm of human chromosome 6 (6p21.3). These discoveries spanned some forty years and laid the foundations of the human MHC as we know it today. Currently
the human MHC is subdivided into three classes: class I, class II and class III. These
subregions and the genes encoded within them will be discussed shortly.
P
h y s ic a l m a p p in g of th e m h cBy the middle of the 1980s, cDNA and genomic clones were available for the HLA-A,
-B and -C class I gene products (for review see Strachan, 1987), the class II DR, DQ
and DP genes (for review see Trowsdale, 1987) and some of the complement gene
products of the MHC class III region (for review see Campbell et a l, 1986). The first
physical map of a region of the MHC was estabhshed in 1986 for the class II region
(Hardy et a l, 1986). Serology and DNA cloning had identified three class II
subregions termed DP, DQ, DR and two additional loci DNA and DOB (Trowsdale et
a l, 1984; Trowsdale and Kelly, 1985 and references therein). Positioning of the
subregions along the short arm of chromosome 6 proved difficult. Family
recombination studies consistently indicated that the DP subregion was centromeric to
DR but a lack of observed recombination between DQ and DR hindered their precise
localisation. Individual subregions were characterised by cosmid cloning but linkage
and the presence of repetitive sequences impeding the cosmid walking. The production
of large restriction fragments combined with pulsed-field gradient gel electrophoresis
(PFGE) was used to counteract the linkage problems (Schwartz and Cantor, 1984; Van
der Ploeg et a l, 1984). This technique allows fragments of DNA up to 2000 kb in size
to be separated by a perpendicularly-orientated and alternately pulsed electric field.
These conditions cause the large DNA coils to elongate parallel to the applied electric
field during electrophoresis. The separation occurs when the alternating field switches
to the perpendicular and the “worm-hke DNA coils” have to reorient themselves to the
new field direction. The time required to reorient is sensitive to the molecular weight of
the DNA fragments and thus a separation according to size is accomplished. The
dinucleotide CG is rare in the human genome occurring at less than one quarter of the
expected frequency. The méthylation of selected cytosine residues is confined to the
dinucleotide CG, creating 5-methylcytosine (5-methyl C), The spontaneous
deamination of nucleotides A, G and C does occur and the products are recognised and
repaired by the appropriate enzymes. However, accidental deamination of 5-methyl C
creates a thymine residue which is indistinguishable from non-mutant T nucleotides and
is not readily recognised by DNA repair mechanisms (Bird, 1987). Therefore C residues can mutate to T over evolutionary time and create a deficiency of CG
dinucleotides. The CG sequences which are present in the vertebrate genome are
clustered into discrete unmethylated ‘islands’ of 1-2 kb, predominantly at the 5’-ends of active genes (Bird, 1987). This phenomenon has been exploited in the identification and
mapping of human genes. Restriction enzymes which recognise CG dinucleotides can
be utilised to cleave genomic DNA at these rare sites, producing large fragments of
DNA for separation by pulsed-field gel electrophoresis. Furthermore, enzymes exist
which will only cut when the CpG is unmethylated, resulting in large fragments that
possibly identify the 5’-ends of genes (Brown and Bird, 1986).
The pulsed-field gel electrophoresis of large fragments created by infrequent-cutting
restriction endonucleases and the subsequent southern blotting and probing of these
fragments with a and p-chain (see structure of class I and class II molecules in this
chapter) probes specific for each of the MHC class subregions resulted in the ordering
of the classical MHC molecules at 6p21.3 (Hardy et a l, 1986; Carrol et a l, 1987).
This information proved invaluable in the cloning of overlapping cosmids spanning
each of the three classes of the MHC (Blanck and Strominger, 1988; Spies et a l, 1988;
Blanck and Strominger, 1990). By combining the physical mapping strategies of long
G
e n e s o f th e h u m a nm h cThe most recent map of the human MHC is shown in Figure L I (Trowsdale and
Campbell, 1997). The MHC is traditionally divided into three classes: most centromeric
on the short arm of chromosome 6 is the class II region, followed by the class III
region and most telomeric is the class I region. The class I and II regions of the MHC
contain numerous genes involved in the processing and presenting of antigens to
T-lymphocytes (these antigen presentation pathways will be described shortly). The
class m region contains genes involved in inflammation, the complement pathway and
heat shock proteins.
Class I genes
The most telomeric region of the MHC contains, amongst others, the classical class I
genes (antigen presenting) HLA-A, -B, -C, -E, -F and -G. There are at present 18
identified HLA class I-related genes and pseudogenes in this region (Geraghty et a l,
1992). Most recently, additional immune-related genes have been identified telomeric
of the traditionally accepted terminus of class I. This has prompted Gruen and
Weissman (1997) to propose that the class I region is expanded by a further 4 Mb
towards the telomere—the so called “extended MHC”. The additional genes include a ubiquitin-like gene, a group of olfactory receptor genes and the Haemochromatosis
gene (Gruen and Weissman, 1997). The localisation of olfactory receptor genes within
the MHC (Fan et a l, 1995) is highly interesting because of their possible involvement
in mate selection (Yamazaki et a l, 1976). Mice are reported to be able to distinguish
potential mates (i.e. non-relatives) on the basis of their MHC haplotypes, through
odorants in urine. It is speculated that the olfactory receptor genes in the class I region
may have a role to play in this mate selection (Potts et a l, 1991). There is some
documented MHC-determined mating-type preference in humans but the reported
findings are highly controversial and tenuous (Wedekind et a l, 1995).
Class III genes
The class III region is the most gene-rich of the MHC classes with over 76 genes
identified to date. Many of the genes are involved in the complement cascade of natural
immunity and some are interferon-inducible heat shock proteins. Most recently a group
of genes involved in inflammation have been identified at the telomeric end of the class
m region. Gruen and Weissman (1997) have proposed that these genes form a distinct cluster and should therefore be classified into the MHC class IV region. Three related
cytokines, TNF, LTA and LTB are found in this region and all have been imphcated in
1995), which is a homologue of the yeast SKI2 (superkiller 2) gene (Widner and
Wickner, 1993). The yeast Ski2 protein exerts an antiviral effect by blocking the
translation of uncapped viral genes (Widner and Wickner, 1993). The role of human
SKI2W has not yet been elucidated. Coincidentally, the closest neighbour of the SKI2
gene in 5. cerevisiae is the RING3-homologue, BDFl (Lygerou et a l, 1994). This is
highly interesting as the RING3 gene is located to the class II region of the MHC in
human, mouse and chicken (see chapters 3-7 of this thesis). Yeast do not have an
MHC, but this close linkage of an antiviral gene with a RING3-homologue may
represent the earliest association of the RING3 gene with an immune-type locus, albeit
a very primitive example.
Class II genes
The class II region is approximately 1 Mb in length and consists of classical and non- classical MHC class n genes. The classical surface-expressed (antigen presenting)
molecules in humans are HLA-DP, -DQ and -DR; the molecules are composed of a and
(3 chains and these genes are arranged as matched pairs i.e. DRA and DRB, DQA and
DQ, DPA and DPB. The number of DRB genes and pseudogenes can vary according to
the haplotype. The DQ and DP regions include a pseudogene pair i.e. DQA2 and
DQB2, DPA2 and DPB2. HLA-DMA and -DMB are linked loci which are distantly
related to classical class II sequences in that they form a molecule which has a and p
domains. Although the products of the DM genes form a heterodimer (HLA-DM) they are not presented on the cell surface but rather catalyse the loading of antigen onto class
II molecules (see class II antigen presentation in this chapter; Denzin and Cresswell,
1995). HLA-DNA and -DOB are non-classical class II genes whose protein products
form a heterodimer called HLA-DO (Jensen, 1998). HLA-DO appears to inhibit the
class n antigen-processing pathway by binding to the protein HLA-DM (Jensen, 1998).
Interestingly, the DNA and DOB loci are not adjacent but are separated by eight other
genes.
A tight cluster of four genes within the class II region is responsible for the processing
of antigens which will ultimately be presented by class I molecules. The TAPI and
TAP2 (transporter associated with antigen processing) genes are members of the ABC
(ATP-binding cassette) transporter superfamily (Townsend and Trowsdale, 1993). The
products of TAPI and TAP2 form a complex in the endoplasmic reticulum (ER)
membrane which is responsible for translocating peptides from the cytoplasm into the
lumen of the ER (Androlewicz and Cresswell, 1994). Once the peptides are transported
to the ER lumen then assembly with the class I molecules can be achieved. LMP2 and
LMP7 are the second pair of genes in this tight cluster which are intimately involved in
antigen processing. They encode components of a large complex called the proteasome
proteasome and are involved in the proteolytic degradation of intracellular and viral
protein antigens which creates specific peptides for presentation to T-lymphocytes.
There are two pseudogenes in the class II region: the IPP-2 (phosphatase inhibitor)
gene (Sanseau et a l, 1994) and a short class I fragment, HLA-Zl, are located between
the LMP2 and DMA loci (Beck et a l, 1996).
The boundaries of the MHC class II region may extend further towards the centromere
than originally thought. This current behef has been confirmed by the discovery of a
cluster of immune-related genes centromeric of DPB2 (Herberg et a l, 1998a). The
Tapasin gene is located within this novel cluster and is responsible for stabilising the
interaction between TAP and class I molecules (Herberg et a l, 1998b; Sadasivan et a l,
1996). Furthermore, approximately 400 kb centomeiic of DPB2 is the BAK gene,
which is a potent inducer of apoptosis (Herberg et a l, 1998c). Evidence suggests that
BAK may be involved in autoimmunity, due to its close location to the MHC. Indeed, BAK is a bcl-2 homologue and the latter has been imphcated in autoimmune
dysfunction in mice with non-obese diabetes (NOD) (Garchon et a l, 1994). The
location of Tapasin so close to the MHC would suggest that the boundaries of the class
II region could be extended. However, this novel cluster of centromeric genes has been
examined and the Alu and G + C content would indicate that the classical class II and
Tapasin regions are contained on separate isochores (Herberg et a l, 1998a). Vertebrate
genomes are non-uniform in gene distribution and base composition, with the G + C
content defining different isochore classes (Bemardi, 1993). A boundary between two
isochores was recently discovered and found to correspond to the boundary between
MHC class II and III regions (Fukagawa et a l, 1995). As the entire MHC is sequenced
and characterised and the genes which neighbour Tapasin are identified, it will be easier
to decide if this new gene is indeed part of an extension of the class II region or if there
is a new class of genes within the MHC.
The RINGS gene is the only expressed gene in the class II region which has no obvious
immunological function. It does not share similarity with any of the classical class II
genes but does show significant homology (up to 80% amino acid identity locally) to
the/5/i gene of Drosophila (Haynes et a l, 1989; Beck et a l, 1992). The RINGS gene
will be introduced more thoroughly later in this chapter
S
t r u c t u r e o f c lass ia n d classn
m h c m o lec u le sClass I and class II MHC molecules are cell surface glycoproteins with similar
structures. These molecules are responsible for presenting pieces of antigen (peptides)
Class I structure
The class I molecules are glycoprotein heterodimers which are classified as type I transmembrane proteins. They are composed of a heavy chain (or a chain) and p2
microglobulin (p2m). There are four external domains, three are contributed by the
heavy chain and are designated a l , a2 and a3 and the fourth is the P2m, a single
domain. The heavy chain is encoded within the MHC by a class I gene whilst p2m,
although showing evidence of a distant evolutionary relationship with MHC molecules,
is located on chromosome 15 in human. Each of the four external domains contains
approximately 90 amino acids and disulphide bonds hold the three a and P2m domains
together. The heavy chain has a 25 amino acid transmembrane region which spans the
membrane as an a helix and the cytoplasmic tail of the heavy chain is composed of
hydrophilic amino acids which form a connection between the intracellular environment
and the cell surface. Class I molecules are expressed by virtually all nucleated cells. The
3-dimensional structure of the class I molecule has been elucidated by X-ray
crystallography (Bjorkman et a l, 1987). This revealed that the folding of the a l and a2
domains created a cleft whose base consisted of antiparallel p sheets. Amino acid residues from the a l and a2 domains, hence of the cleft, were shown to form the
peptide binding region (PER), for the presentation of antigen to T-cells. The high
polymorphism or hypervariability of the PER allows an enormous number of different
antigens to be presented to the immune system.
Class II structure
Class II MHC molecules are also glycoprotein heterodimers which are composed of a
and p heavy chains, all of which are encoded within the MHC. The a chain is
composed of two extracellular domains, a l and a2; likewise, the p chain has domains
pi and p2. The a l and pi domains are situated distal to the cell membrane, whilst the
a l and p2 domains are proximal to the cell membrane. Class II molecules have
transmembrane-spanning regions, one from each of the a and p chains, which are
believed to cross the cell membrane as a-helices. The hydrophobic cytoplasmic tails of
the a and p chains are 10-15 residues in length. The a l and pi domains contain highly
polymorphic amino acids and fold to generate a cleft which becomes the PER of the
class n molecules (Erown et a l, 1993). The class II cleft stmcture is similar to the class
I cleft but the shape of the groove, generated by the folding, is open-ended for class II
and closed for class I molecules. This difference allows discrimination in the binding of
peptides; class It molecules can bind longer peptides (typically 12 to 24 residues) than
class I molecules. Class II molecules are expressed primarily on E lymphocytes, macrophages, dendritic cells and activated T lymphocytes; all are loosely termed antigen
MHC CLASS I AND CLASS H GENE STRUCTURE
Human class I and II genes are classified as members of the immunoglobulin (Ig) gene
family (Klein, 1986). The class I genes of different species are very similar and tend to
be quite small (Trowsdale, 1995). The first exon generally contains the 5’-untranslated
(UTR) region and the signal sequence. This is followed by three exons which encode
the a l , a2 and a3 external domains. The fifth exon encodes the transmembrane domain
and the last two or three exons contain the cytoplasmic domains and the 3’-UTR
(Trowsdale, 1995).
The class II genes do show some variation in gene structure but in mammals the A (a)
chain genes usually have 5 exons and the B (p) chain genes have 6. The basic
organisation is as follows: an exon for the 5’-UTR and signal sequence, two separate
exons for the a l , a2 or p i, p2, an exon containing the connecting peptide,
transmembrane and cytoplasmic domains and a final exon which contains the 3 -UTR
(Trowsdale, 1993).
A
n t ig e n p r e se n t a t io nby
c lassi
a n d c lassn
m o l e c u l e sThe class I and II molecules are responsible for binding peptides (antigens) and
presenting them to T-cells, thereby ilhciting an adaptive immune response. The structures of these two classes of molecules are different, although related (as
previously described) and they are involved in different pathways for the presentation
of antigens.
Class I antigen presentation (for a review see Monaco, 1992)
Peptides which are loaded into class I molecules are primarily derived from an
endogenous (intracellular) source. This usually equates to cells which are infected with
viral particles or bacteria and cells which have been geneticaUy altered, i.e. cancerous
cells. This is important because the mechanism of class I antigen presentation usually
results in the cytotoxic kiUing of the infected cells. Class I molecules can occassionally
present exogenous peptide but this wiU not be discussed here. The proteasome, of
which the gene products of LMP2 and LMP7 are subunits, degrades cytoplasmic
proteins in the cytosol (Goettrup et a l, 1996). One model suggests that the LMP
complex may have multiple catalytic sites for the simultaneous production of multiple
peptides. Class I molecules preferentially bind peptides of 9 (±1) amino acids
(nonamers); nonameric peptides bind to class I molecules with an affinity 100-1000-
fold greater than longer or shorter peptides (Schumacher et a l, 1991). The LMP
complex is involved in generating such nonamers (Monaco, 1992). There is evidence to
little competition from nonameric peptides (Monaco, 1992). The generated peptides are
transported into the endoplasmic reticulum (ER) by the heterodimerc TAP1/TAP2
molecule which is a member of the ATP-binding cassette (ABC) family of transporters.
The TAP1/TAP2 molecule spans the ER membrane. Once in the ER lumen, the peptide
loading of class I molecules is regulated by several chaperones. Free MHC class I
heavy (a chain) chain binds with calnexin (Williams and Watts, 1995). In humans,
calnexin is released when the (32m molecule binds to the heavy chain; caketiculin is the
chaperone at this stage (Sadasivan et a l, 1996). The heavy chain-(32m-calreticuhn
complex then interacts with the TAP1/TAP2 molecules, to allow the loading of peptide
into the class I molecule. This interaction of class I molecule and TAP is stabilised by
the glycoprotein Tapasin (Sadasivan et a l, 1996). Once bound, the heterotrimeric
class I molecule (heavy chain-(32m-peptide) proceeds to the cell surface via the Golgi
apparatus (Koopman et a l, 1997). At the cell surface the class I molecule presents the
bound peptide to activated T cells which have the CDS surface antigen. These T cells
are often called cytotoxic T lymphocytes (CTLs) and they recognise specific class I- bound peptides; this recognition is dependant upon the specificity of the T cell receptor
(TCR). If the TCR recognises and binds with the presented peptide, an immune
response is initiated which generally results in the lysis of the cell which was presenting
the peptide.
C lass II antigen p resen tatio n (for review see Neefjes and Ploegh, 1992 and
Pieters, 1997)
Peptides bound to MHC class II molecules are predominantly derived from exogenous
(extracellular) antigen. Class II molecules are only found on so-called “antigen
presenting cells” (APC) e.g. macrophages, B cells, dendritic cells and activated T cells.
These cells internalise antigen by receptor-mediated endocytosis and phagocytosis and
deliver it to the intracellularly-located endosomes and lysosomes. These organelles
degrade the internalised antigen into peptides of approximately 12-24 residues in
length. Class II molecules can accommodate a wider range of peptide length than class I
molecules, because the PBR is open at one end (Brown et a l, 1993).
MHC class II molecules are assembled in the endoplasmic reticulum (ER) as a and p
heterodimers and are stabilised by a membrane-bound chaperone protein called the
MHC class n-associated invariant chain or y chain. A segment of the y chain appears to
act as a surrogate peptide by binding directly with the peptide-binding region. Class II
molecules exit the ER as a complex of three y chains with three assembled ap dimers
and are transported to the trans-Golgi reticulum. At this destination, the class II
molecules are directed to the endocytic pathway by the y chain which is then rapidly
degraded by proteases. Only a small fragment of the y chain remains protected from
invariant chain peptide) and it is only displaced when a peptide is ready to be bound.
The displacement of CLIP is catalysed by HLA-DM, the product of the DMA and DMB
genes, and the class II molecule can then bind “authentic” peptide (Jensen, 1998). The
acidic pH in endosomes and lysosomes is thought to contribute towards the efficient
degradation of the antigens and the resulting binding of the generated peptides to class
n molecules. Once peptide is bound to the ap heterodimer, the complex is transported
to the cell surface where it is recognised by a specific T lymphocytes carrying the CD4
surface antigen. Consequently an adaptive immune response is initiated. MHC class II
molecules can occassionally present peptides derived from endogenous antigen.
Autophagy is a mechanism which allows the engulfment of cytosohc material to create
autophagosomes. These structures fuse with lysosomes and the originally endogenous
antigen is thus delivered into the endocytic pathway for eventual binding with class II
molecules. Thus the class II antigen processing and presenting pathway is capable of
utilising both exogenous and endogenous antigens.
C
o m pa r a t iv e g e n o m ic s a n d e v o l u t io n of th e m h cThe arrangement of class I, II and
in
genes within the MHC appears to have beenbroadly conserved throughout mammals (Trowsdale, 1995). The mouse MHC is very
well defined and is organised much the same as the human MHC. In mouse the
complex is called H-2 and is found on chromosome 17. Further details of the
mouse/human comparative map are discussed in chapter 4. Rodents and ancestral
primates are estimated to have diverged from a common ancestor 80-100 million years
ago, thus comparison of mouse and human MHCs can be helpful in deciphering the
MHC map of their common ancestor.
In the MHC class I region it would appear that several independent rounds of
replication occurred after the separation of mouse and human lineages. Consequently,
human class I genes are more similar to each other than to the mouse class I loci ( Ahnini
et a l, 1997). The genes have species-specific characteristics in terms of sequence and
position, which have masked their orthologous (relationships of homology by descent
from a common ancestor without duplication) origins. Class I genes in mammals appear
to be rapidly tumed-over i.e. some loci are lost by silencing and/or deletion, whilst new
genes evolve by gene duplication (Hughes and Yeager, 1997). Furthermore, the class I
H2-K genes (H2-K and H2-K2) are positioned at the proximal end of the mouse class n region; this is a direct insertion of some 60 kb of DNA compared to the human MHC
map (Hanson and Trowsdale, 1991). The class II region shows a high degree of
homologues are also found in the mouse in equivalent positions (see chapter 4), The
different subfamilies appear to have undergone independent duplication in human and
mouse. The class
in
region is highly conserved between the two species and mostgenes have an orthologous relationship (Gasser et a l, 1994).
The early evolutionary history of the immune system remains obscure but phylogenetic
analysis of sequence data generated from mammals has allowed some evolutionary
mechanisms to be elucidated. The MHC class H subregion appears to have arisen by
gene duphcation prior to the divergence of placental mammals, whilst the class I region
displays continuous rounds of gene duplication throughout the evolution of the MHC
(Hughes and Yeager, 1997). In protein-coding regions, mutations that cause changes in
the amino acid sequence (nonsynonymous mutations) are generally deleterious and are
rapidly eliminated from the population by natural selection. Mutations in coding regions
that do not result in an amino acid change (synonymous mutations) are regarded as
selectively neutral and may be lost or become fixed in the population. This prediction is
supported for the coding regions of many genes. However, if natural selection were acting to favour diversity at the protein level, then the number of nonsynonymous
nucleotide substitutions would be expected to exceed the number of synonymous
substitutions. Only a few examples of this pattem have ever been identified and MHC
genes have been implicated. Both class I and class II genes display a very high level of
polymorphism in human and mouse. The reason for the polymorphism was enigmatic until the function of MHC molecules was elucidated; different aUehc products at the
MHC loci can differ in the spectmm of antigens which they can bind and present to T
cells (Doherty and Zinkemagel, 1975). Doherty and Zinkemagel argued that this
function of MHC molecules would favour heterozygote advantage (or overdominant
selection) i.e. an individual who was heterozygote at all or the majority of MHC loci
would have an advantage over a homozygote, because the former would be able to bind
a wider range of peptides and thus resist a wider range of pathogens. Hughes and Nei
(1988) examined the amino acid sequence of class I molecules at the peptide binding
region (PER) and found that the number of nonsynonymous substitutions exceeded
synonymous substitutions i.e. overdominant selection at the PER. When they examined
the regions outside of the PER, the number of synonymous changes was greater than
the nonsynonymous amino acid changes (Hughes and Nei, 1988), as is the case for
most genes. A similar pattem was found when the class II molecules were examined
(Hughes and Nei, 1989). Other authors disagree with the high polymorphism of MHC
molecules being driven by heterozygote advantage in the resistance to pathogens.
Disease associations with the human MHC are mainly autoimmune by nature and there are only a few examples of a particular MHC haplotype being associated with resistance
to a disease or pathogen. One HLA haplotype confers resistance to a phase of acute
class n gene rather than a closely-linked but unrelated gene. Furthermore, this
association is not very strong (Hill gfaA ,1991). Kaufman and co-workers (1995) have
described three alternative explanations for the high polymorphism, “accumulating,
merging and boosting”. The first explanation is that there are a large number of alleles
in the MHC because they accumulate over the course of evolution and do not disappear
quickly. They may once have been under selection, as evidenced by the greater number
of nonsynonymous mutations relative to synonymous changes in the PER, but the
different alleles may not be under continuous selection. The second explanation is that
many small, isolated populations each had a different set of polymorphic MHC genes which arose by selection and genetic drift in the small isolates. The individual isolates
merged, thereby giving rise to highly polymorphic MHC alleles. The third explanation
suggests that there may be another use for MHC alleles besides the resistance to
pathogens or that the alleles are very closely linked to other genes which are selected for
high polymorphism. The two loci are proposed to “hitch-hike” with each other and the MHC genes are said to be “boosted” by the linked genes to give high polymorphism.
Mate selection, recognition of kin and success in reproduction may depend upon MHC
alleles and in turn be causing the high polymorphism (Potts and Wakeland, 1993).
The origin of the high polymorphism remains controversial and fiercely debated, as
does the question of the MHC functioning as a cluster of genes. There are genes in the
MHC which do not have an obvious immune function and many of these are located in
the class III region. As this region is positioned between class I and II in human and mouse, it has been suggested that class III was inserted as a block by a transposition
event (Trowsdale, 1995). Class I and II are located close to each other in chicken and
rabbits (Trowsdale, 1995). The class I and II genes may have diverged from a common
ancestor over 700 million years ago and many duplications have occurred during the
evolutionary history of the MHC. The MHC functioning as a gene cluster is most
pronounced in the class II region, where the class I antigen processing (LMP) and
antigen transporting (TAP) genes are found with the antigen presenting class II genes.
It is still unclear if the TAPs and LMPs were recruited into the MHC or if they arose in
the MHC de novo. Certainly LMP2 and LMP7 have homologues, Ô and M Bl (or X)
respectively, which are located on other chromosomes and are constituitively expressed
(Belich et a l, 1994). Phylogenetic analysis of LMP7 and MBl (X) estimates that these
two genes diverged around 600 million years ago, prior to the divergence of jawed and
jawless vertebrates (Hughes and Yeager, 1997). This may suggest that the origin of the
class I MHC could be as old since LMP7 is extremely important for correct antigen
processing in MHC class I antigen presentation (Hughes and Yeager, 1997). Most
recently, Kasahara and co-workers (1996) have described the finding of a region on chromosome 9 which contains numerous MHC homologues. They speculated that the
LINKAGE DISEQUILIBRIUM AND THE MHC
The human MHC region displays significant linkage disequilibrium or non-random
association between particular pairs of alleles at different loci. If alleles associate randomly then all pairwise combinations would be observed in a population at equal frequencies. However, some allele combinations are observed together more often than expected and others are rarely associated at all. This phenomenon of population genetics is called linkage disequilibrium and its value, D, is the difference between the observed frequency and the frequency expected under random association of the alleles.
A good example of linkage disequilibrium in Caucasian populations is observed in the HLA alleles A l, B8 and DR3 (Thomson, 1995). Extremely high disequilibrium is observed between A l and B8 in North European populations. Alleles B8 and DR3 are also observed together at higher than expected frequencies. Several factors can create linkage
disequilibrium: selection (direct or hitchhiking); migration and admixture; the occurrence of a new mutation and sampling or drift effects. Linkage disequilibrium does not have to be formed only between linked loci. It can be created between loosely linked or completely unlinked loci by, for example, strong selection or the recent admixture of two populations with very different allele frequencies at the loci under study. If selection is weak or removed, linkage disequilibrium decreases as a linear function with each generation as recombination occurs between the loci. Therefore, for unlinked loci linkage disequilibrium decreases rapidly, but for tightly linked loci it can be maintained for very many generations.
If a genetic marker shows association with a disease, the disease susceptibility could be directly influenced by the presence of the marker alleles. Alternatively, the marker allele association with the disease could result from linkage disequilibrium between the marker allele and a disease-predisposing locus. The gene which causes hereditary haemochromatosis (HH) - an autosomal recessive disorder of iron metabolism - was initially linked to the MHC
region by observing linkage disequilibrium of the HLA-A3 allele in HH patients (Feder et a l,
1996). The gene was finally located to the “extended MHC region”, a region telomeric of the MHC class 1 region. There are numerous diseases which show associations with HLA
Yeager (1997) disagree with this finding and propose that the genes involved in the
duplication occurred at different times, “spread over at least 1.6 billion years”. Further
discussion of this duplication is presented in Chapters 5 and 6.
The evolution of the MHC is very complex and only when aU of the genes have been
elucidated will it be possible to finally begin to understand how the different genes
originated and diverged.
S
e q u e n c in g o f th e h u m a n m a jo r h ist o c o m p a t ib il it yCOMPLEX
A combined effort by several groups enabled the cloning of the entire human MHC
class II and HI regions into overlapping cosmids (reviewed by Campbell, 1993) and the
entire MHC has now been cloned into yeast artificial chromosomes (Abderrahim et a l,
1994). The availability of these clones made possible the systematic effort to sequence
and characterise the human MHC and indeed approximately 40% (350 kb) of the class
n region had been completed by 1995 (Beck et a l, 1992a; Radley et a l, 1994; Beck et
a l, 1996). In September of 1996, the systematic sequencing of chromosome 6 began at
the Sanger Centre in close collaboration with the chromosome 6 community. The MHC
was the most advanced region of chromosome 6, in sequence and characterisation
terms, because of the intense interest in the genetics and biology of this gene cluster. It
was therefore given the highest priority for sequencing completion and was divided
between five groups:
• Class I - Hidetoshi Inoko, Tokai University, Japan and Dan Geraghty, University
of Washington, U.S.A.
• Class II - Stephan Beck, The Sanger Centre, U.K., in collaboration with John
Trowsdale, Cambridge University, U.K.
• Class m - Duncan Campbell, MRC Oxford, U.K. and Leroy Hood, University of
Washington, U.S.A.
The Sanger Centre effort has to date resulted in approximately 90 % (0.7 Mb) of the
MHC class II being completely sequenced. There are 18 expressed genes (from
centromere to telomere): DPBl, DPAl, DNA, RING3, DMA, DMB, LMP2, TAPI,
LMP7, TAP2, DOB, DQBl, DQAl, DRB1, DRB2, DRB3, DRA, BTN (Beck et a l,
1996; Avis e ta l, 1997). Additionally, 12 pseudogenes (HLA-Zl, IPP2, DQA2,
DQB2, DQB3, RINGS, -9, -13, -14, DRB7, DRB8 and DRB9) have been identified
M
a p p in g a n d c lo n in g of th e h u m a n r in g s c d naCosmid cloning of the MHC extended into the class II region and linkage between
cosmids was established by chromosome walking; the systematic screening of genomic
libraries with unique probes to identify overlapping cosmids. A 120 kb chromosome
walk extended from the DNA gene in the 3’ direction and isolated 6 cosmids between
DNA and the DMB genes (Blanck and Strominger, 1990). Efforts were made to locate
cross-hybridising a and p chains in the DNA cluster but this was unsuccessful. The
authors suggested that there were no additional class II genes neighbouring DNA and
they made the cosmids publically available.
The tendency of CpG islands to cluster at the 5’-end of genes was exploited in the
identification of novel MHC class II genes. “Rare-cutter” restriction endonucleases that
recognise sequences containing one or more unmethylated CpG dinucleotides, were
utilised to identify these CpG islands, PFGE blots prepared from the cleavage of
human genomic DNA with combinations of rare-cutter endonucleases, were
sequentially hybridised with probes specific for all of the classical class II loci, HLA-
DP, -DQ, -DR, -DNA and -DOB (Hanson et a l, 1991). Combining these results with
published physical mapping data positioned four clusters of rare-cutter sites within the
class n region; two occurred between the HLA-DNA and HLA-DOB genes. Cloning
of the four CpG clusters involved extensions of previously published cosmid walks and the utilisation of chromosome jumping. Once the most centromeric unmethylated rare-
cutting cluster had been cloned then the adjacent cluster was isolated by using rare-
cutter jumping libraries (Poustka and Lehrach, 1991). Briefly, loci positioned several
kilobases apart in the genome can be subcloned next to each other by restriction
digestion with infrequent-cutting endonucleases so that the ends of the fragments
contain the two loci of interest. Re-circularisation (intramolecular ligation) of these large
fragments brings the loci of interest to adjacent sites; the loci could chromosomally be
separated by several hundred kilobases. Complete or partial cleavage of these circles
will then release the fragments containing the junction between the two ends and they
are generally small enough to be cloned into a lambda insertion vector. A probe
originally specific for only one of the loci can be used to isolate the neighbouring locus
in a jumping library because of the reduced distances between them. This technology
was utilised to “jump” from the first CpG cluster to the second cluster between DNA
and DOB (Hanson e ta l, 1991). To identify potential coding sequences, zoo blot cross
hybridisation and Northern blot analyses were performed with human probes specific to
the CpG cluster. The consensus being that transcribed DNA sequences tend to be more
highly conserved in evolution and thus a transcript identified in several species and/or
tissues would be highly indicative of a gene. This cluster was found to contain