_____________________________________________________________________________________________________
*Corresponding author: E-mail: [email protected];
22(4): 1-11, 2018; Article no.CSIJ.41427
ISSN: 2456-706X
(Past name: American Chemical Science Journal, Past ISSN: 2249-0205)
Theoretical Study of the Chemical Reactivity of Five
Schiff Bases Derived From Dapsone by the DFT
Method
Jean Stéphane N’dri
1, Mamadou Guy-Richard Koné
1*,
Charles Guillaume Kodjo
1,2,
Ahmont Landry Claude kablan
3, Sopi Thomas Affi
1,
Lamoussa Ouattara
1and Nahossé Ziao
11
Laboratoire de Thermodynamique et de Physico-Chimie du Milieu, UFR SFA, Université Nangui Abrogoua, 02 BP 801 Abidjan 02, Côte-d’Ivoire.
2Laboratoire de Chimie BioOrganique et de Substances Naturelles, Université Nangui Abrogoua,
UFR-SFA, 02 B.P. 801 Abidjan 02, Côte-d’Ivoire.
3
UFR des Sciences Biologiques, Université Péléforo Gon Coulibaly de Korhogo, BP 1328 Korhogo, Côte d’Ivoire.
Authors’ contributions
This work was carried out in collaboration between all authors. Authors JSN and MGRK designed the study, performed the statistical analysis, wrote the protocol and first draft of the manuscript. Authors CGK and NZ managed the analyses of the study. Authors ALCK, STA and LO managed the literature searches. All authors read and approved the final manuscript.
Article Information
DOI: 10.9734/CSJI/2018/41427
Editor(s):
(1)Gustaaf Schoukens, Professor, Department of Textiles, Ghent University, Belgium.
Reviewers:
(1)Ahmed Mohammed Abu-Dief Mohammed, Sohag University, Egypt. (2)Sema Salgin, Cumhuriyet University, Turkey. (3)Daniel Glossman-Mitnik, CIMAV, Mexico. Complete Peer review History:http://www.sciencedomain.org/review-history/24546
Received 19th February 2018 Accepted 30th April 2018
Published 9th May 2018
ABSTRACT
This theoretical study of chemical reactivity was conducted using the method of the Density Functional Theory (DFT), with B3LYP/6-311G (d, p) as calculation level. A series of five (05) Schiff bases derived from 4.4 '-diaminodiphenylsulfone (Dapsone) was concerned and allowed to predict the chemical reactivity of these compounds. Molecular geometries and electronic properties such as the energies of the frontier molecular orbitals (HOMO and LUMO), ionization potential (I), and
electron affinity (A) were examined to obtain a better overview of the molecular properties. Thus compound 4, whose energy gap between the frontier orbitals (HOMO and LUMO) is ΔEgap = 3.910 eV appears to be the most polarizable. It is the most reactive with the lowest kinetic stability compared to all the studied molecules. The values of the global reactivity descriptors confirmed the great chemical reactivity of the compound 4. Local indices of reactivity and dual descriptors were calculated to indicate the probable sites of Electrophilic and nucleophilic attacks of the various studied compounds.
Keywords: Chemical reactivity; global descriptors; local descriptors; dual descriptors.
1. INTRODUCTION
The fight against infectious diseases remains a challenge of public health essential; this is explained by the high mortality and morbidity rate linked to these diseases [1,2]. Also, the anti-infectious chemotherapies are now losing
efficacy because of drug-resistance’s
phenomenon of infectious germs [3]. Nowadays Dapsone presents many undesirable effects such as cutaneous, neurological and psychiatric infections. It shows resistances during treatment too [4]. In such context, the continuation of the development of new effective drugs becomes a real necessity. Several researchers have recently set up Schiff bases and azetidinones derived from Dapsone [5-7]. In addition, the Quantitative Structure-activity Relationship (QSAR) [8, 9] studies of this series were made in order to understand well their chemical reactivity. With the development of computational techniques and computational chemistry, quantum chemistry provides an overview of the electronic structures
of molecules and strongly propels the
development of the traditionally experimental chemistry [10]. Currently, the DFT method has been accepted as a popular approach for
calculating the structural characteristics and energies of molecules by the scientific community [11-14] and for the efficacy and
accuracy of evaluating a number of molecular properties [15]. Parr and Yang
followed the idea that well-known chemical properties such as electro-negativity, chemical potentials and affinities could be accurately described and computed by manipulating electron density as the fundamental quantity [16, 17]. On the other hand, from the work of Fukui and its theory of Frontier molecular orbitals (FMO) [18], The same authors have generalized
the concept and proposed the function of Fukui as a tool for describing local reactivity in molecules [19,20]. The present study focuses on five (05) Schiff bases derived from
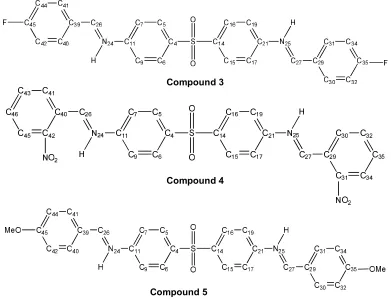
Dapsone that are shown in Fig. 1.
These Schiff bases are obtained by
condensation of Dapsone with benzaldehydes
diversely substituted in ethanol in the presence of the concentrated hydrochloric acid
as a catalyst, were synthesized and tested by S. J. Wadher et al. [7] for their antibacterial and antifungal activities. These molecules presented millimeters. The purpose of this work is to
Compound 1
Fig. 1. Molecular structures and numbering of Schiff’s atoms bases derived from Dapsone
determine theoretically the privileged sites of electrophilic and nucleophilic attacks on the aromatic carbon atoms contained in these Schiff bases, by different quantum methods.
2. MATERIALS AND METHODS
2.1Level of Calculation
The theoretical study of chemical reactivity was conducted on the basis of three theoretical approaches. The first concerns the analysis of surfaces of Molecular Electrostatic Potential (MEP).The second approach is relative to the Frontier Molecular Orbitals (FMO). The latest approach focuses on local indices of reactivity as well as dual descriptors. The geometries of the molecules have been optimized at the DFT calculation level with the functional B3LYP [21,22] in the base 6-311 G (d, p) using the software Gaussian 09 [23]. This hybrid function gives better energies and is in good agreement with ab initio high-level methods [24,25]. Concerning split-Valence and Triple-zeta (6-311G (d, p)) base, it is sufficiently extended and the taking into account of polarization functions
are is important for the explanation of the pairs of electrons of heteroatoms those are not involved in a bound. The geometries are (kept) maintained constant for cationic and anionic systems. Global reactivity indices were obtained from the conceptual DFT model [16]. The
Hierarchical Cluster Analysis (HCA) was
conducted using XLSTAT software [26]. For chemical reactivity indices, they were determined by using electronic populations calculated with the Mulliken Population Analysis (MPA) [27].
2.2 Reactivity Descriptors
2.2.1 Global Descriptors
To predict chemical reactivity, some theoretical descriptors related to the conceptual DFT have been determined. In particular, the Lowest Unoccupied (vacant) Molecular Orbital (LUMO) energy (ELUMO), the Highest Occupied Molecular
Orbital (HOMO) energy (EHOMO), the
electronegativity (χ), the global softness (σ), and the global electrophilicity Index (ω). These descriptors are all determined from the optimized molecules. It should be noted that, the Compound 3
Compound 4
descriptors linked to the frontier molecular orbitals have been calculated in a very simple
way according to the approximation of
Koopmans [28].The LUMO energy characterizes the sensitivity of the molecule to nucleophilic
attack, and concerning HOMO’s one, it
characterizes the susceptibility of a molecule to an electrophilic attack. The electronegativity (χ) is the parameter that translates the ability of a molecule not to let its electrons escape. The global softness (σ) expresses the resistance of a system to the change in its number of electrons. The global electrophilicity index characterizes the electrophilic power of the molecule. These different parameters are calculated from the following equations (1):
= − = −
= − = − 1 2⁄ ( + )
= ( − ) 2⁄ (1)
= 2 = 1⁄
2.2.2 Local descriptors and dual descriptors
In order to differentiate the reactive behaviors of atoms forming a molecule, different indices were used. These are precisely local indices and dual descriptors of reactivity. Local reactivity descriptors, such as function of Fukui [29]
( , ) , local softness ( , ) , local
electrophilic power ( , ), and dual descriptors were proposed to explain the selectivity of electrophilic and nucleophilic attacks of the molecule. It should be recalled (remembered) that the function of Fukui expresses reactivity when the molecule is attacked by a nucleophilic
reagent, whereas the function of
Fukui provides information about the
electrophilic attack on a given site. The value of the highest Fukui function is assigned to the most active site. The condensed indices and express the ability of a site to receive electron density by nucleophilic attack, in
contrast, the and indices express the
ability of a site to yield electron density by electrophilic attack. With regard to the dual descriptor, it is a good tool to predict reactivity and to understand the problem of regioselectivity. Indeed, a dual positive descriptor corresponds to a site which is able to receive electron density, in other words, it is the most electrophilic.
Conversely, a dual negative descriptor
corresponds to a site which is capable to yield
electron density, so it is the most nucleophilic. A site with a value of the dual descriptor close to zero corresponds to a site whose capacity to receive and to yield electron density are equivalent. The different values of the local descriptors are calculated from the equations (2) below [30-33]:
= ( + 1) − ( )
= ( ) − ( − 1)
=
= (2) =
=
Where: ( ): Electron population of the atom k in the neutral molecule
( + 1): Electron population of the atom k in the anionic molecule.
( − 1): Electron population of the atom k in the anionic molecule.
The values of the dual descriptors [33-36] are obtained from the equations (3):
∆ = −
∆ = − (3)
∆ = −
3. RESULTS AND DISCUSSION
3.1 Molecular Electrostatic Potentials (MEP)
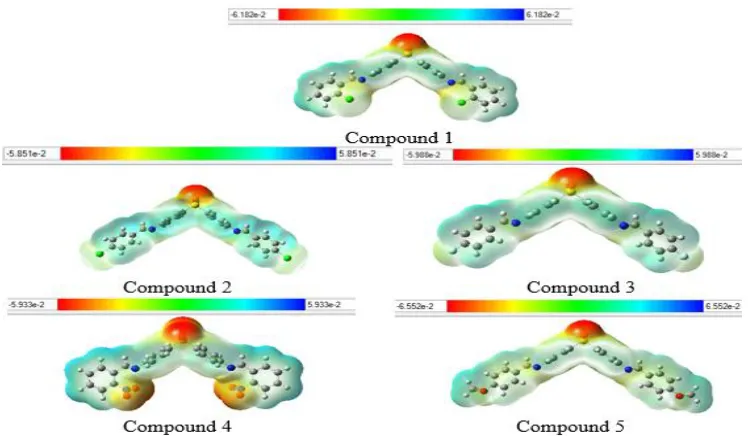
The surfaces of the electrostatic potentials of studied molecules were drawn after optimization at the level B3LYP/ 6-311G (d,p). They are shown in Fig. 2.These surfaces are associated with a color code which is moving continuously from red indicating the most negative potentials to blue indicating the most positive potentials [37] passing through orange, yellow and green [38].
These maps indicate, on the one hand, that the heteroatoms (S, O, N, Cl et F) are the sites of electrophilic attacks because they have a red proximity area of negative potential with a high electron concentration around the (-SO2-) bond of the Dapsone nucleus. On the other hand, the carbon atoms, except those involved in the
azomethine binding (-CH=N-), are more
Fig. 2. Surfaces of Molecular Electrostatic Potentials of Schiff bases derived from Dapsone
3.2 Analysis of Frontier Molecular Orbitals
The Highest Occupied Molecular Orbital (HOMO) and Lowest Unoccupied Molecular Orbital (LUMO) called frontier orbitals and they play a fundamental role in the qualitative interpretation of chemical reactivity [39]. The highest occupied molecular orbital (HOMO), which can be considered as the external orbital containing electron, tends to give these electrons as an electron donor. On the other hand, the lowest
unoccupied molecular orbital (LUMO) is
perceived as the lowest orbital containing free places so able to accept electrons [40]. Therefore, while the energy of HOMO is directly related to the ionization potential, that of LUMO is directly related to electron affinity. The energy difference between HOMO and LUMO called Energy Gap, is an important stability factor for the structures [41]. The HOMO-LUMO energy gap helps to characterize the chemical reactivity
and kinetic stability of the molecule [42]. A molecule with a high energy gap (ΔEgap) is less
polarizable and is generally associated with low chemical reactivity and high kinetic stability
[43]. Fig. 3 below represents the molecular orbital of HOMO and LUMO of Dapsone derivatives obtained using the method B3LYP/6-311 G(d,p).
The energetic parameters of the frontier orbitals are gathered in Table 1.
These results show that compound 4 has the smallest energy gap (ΔEgap = 3.910 eV). So it is the most polarizable and possess the highest chemical reactivity and the lowest kinetic stability among all the studied molecules. On the other hand, compound 3 has the greatest value of the energy gap which is 4.154 eV. Thus, molecule 3 is the least polarizable, with low chemical reactivity and high kinetic stability among the five (05) studied molecules.
Table 1. Energetic descriptors of the studied compounds
Compounds EHOMO(eV) ELUMO(eV) ΔEgap (eV) I (eV) A (eV)
1 -6.438 -2,380 4.058 6.438 2.380
2 -6.573 -2.492 4.081 6.573 2.492
3 -6.500 -2.346 4.154 6.500 2.346
4 -6.671 -2.761 3.910 6.671 2.761
5 -6.083 -2.018 4.066 6.083 2.018
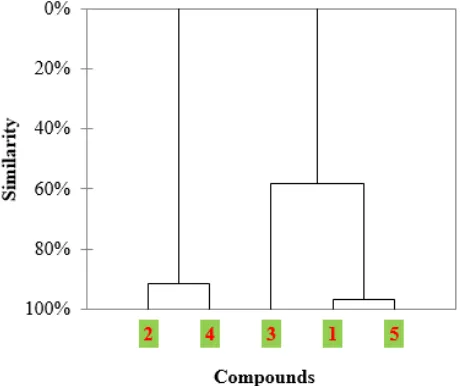
3.3 Hierarchical Cluster Analysis (HCA)
Fig. 4. shows the HCA of the studied molecules. The horizontal lines represent the compounds. The vertical lines the similarity values between pairs of compounds, a compound and a group of compounds and among groups of compounds.
We can note from this analysis that the studied compounds were grouped into three categories: the most active compounds are 2 and 4, compound 3, moderately active and less active compounds 1 and 5.
3.4 Reactivity Descriptors
3.4.1 Global reactivity descriptors
The study of the global reactivity of molecules is based on the calculation of the global indices inferred from the electronic properties. The global indices of the reactivity of the studied Schiff bases were calculated from the equations (1) and recorded in Table 2.
The chemical hardness (softness) value of compound 4 (η = 1.955 eV) is the lowest among studied molecules. Thus, it appears that
compound 4 is more reactive than all the studied compounds. Also, we note that compound 4 has its value of electronegativity (χ = 4.716 eV) which is higher than other compounds' value so it is the best electron acceptor. In addition, the electrophilic index value of compound 4 (ω = 5.688 eV) indicates that it is the most electrophilic one.
3.4.2 Local and dual reactivity descriptors
Local indices and dual descriptors of reactivity were also determined for each molecule according to equations (2). The concerned sites are the carbon atoms linked to a hydrogen atom in the different aromatic nuclei of each molecule. These different indices and descriptors of reactivity are grouped in Tables 3-7.
The values of the local and dual descriptors of compound 1 calculated at the level B3LYP/ 6 311G(d,p), show that the carbon atoms C9 and C19with the value Δω = -0.070, are the preferred sites of electrophilic attack. According to this same level of calculation, a nucleophilic attack will take place preferentially on the C35 and C46 atoms.
Table 2. Global descriptors for chemical reactivity of Schiff bases 1-5
Compounds μ (eV) χ (eV) η (eV) σ (eV) ω (eV)
1 -4.409 4.409 2.029 0.493 4.790
2 -4.532 4.532 2.040 0.490 5.034
3 -4.423 4.423 2.077 0.481 4.709
4 -4.716 4.716 1.955 0.511 5.688
5 -4.050 4.050 2.033 0.492 4.035
Table 3. Reactivity descriptors of compound 1 computed using Mulliken population analysis (MPA)
Sites Local descriptors Dual descriptors
f- f+ σ- σ+ η- η+ ω- ω+ Δf Δσ Δω
C5 0.008 0.011 0.004 0.005 0.017 0.022 0.039 0.051 0.002 0.001 0.012
C6 0.010 0.012 0.005 0.006 0.020 0.024 0.047 0.057 0.002 0.001 0.010
C7 0.021 0.024 0.010 0.012 0.042 0.048 0.099 0.114 0.003 0.001 0.014
C9 0.033 0.019 0.016 0.009 0.068 0.038 0.160 0.090 -0.015 -0.007 -0.070
C15 0.008 0.011 0.004 0.005 0.017 0.022 0.039 0.051 0.002 0.001 0.011
C16 0.010 0.012 0.005 0.006 0.020 0.024 0.047 0.058 0.002 0.001 0.011
C17 0.021 0.024 0.010 0.012 0.042 0.048 0.099 0.114 0.003 0.001 0.014
C19 0.033 0.019 0.016 0.009 0.068 0.038 0.160 0.090 -0.015 -0.007 -0.070
C30 0.018 0.021 0.009 0.010 0.036 0.043 0.086 0.101 0.003 0.002 0.015
C32 0.008 0.009 0.004 0.004 0.015 0.017 0.036 0.041 0.001 0.000 0.005
C34 0.010 0.006 0.005 0.003 0.021 0.013 0.050 0.030 -0.004 -0.002 -0.020
C35 0.017 0.032 0.008 0.016 0.035 0.064 0.082 0.152 0.015 0.007 0.070
C41 0.018 0.021 0.009 0.010 0.036 0.043 0.086 0.101 0.003 0.002 0.015
C43 0.008 0.009 0.004 0.004 0.015 0.017 0.036 0.041 0.001 0.000 0.005
C45 0.010 0.006 0.005 0.003 0.021 0.013 0.050 0.030 -0.004 -0.002 -0.020
C46 0.017 0.032 0.008 0.016 0.035 0.064 0.082 0.152 0.015 0.007 0.070
Table 4 . Reactivity descriptors of compound 2 computed using Mulliken population analysis (MPA)
Sites Local descriptors Dual descriptors
f- f+ σ- σ+ η- η+ ω- ω+ Δf Δσ Δω
C5 0.010 0.010 0.005 0.005 0.021 0.021 0.053 0.052 0.000 0.000 -0.001
C6 0.010 0.014 0.005 0.007 0.021 0.028 0.051 0.070 0.004 0.002 0.020
C7 0.020 0.024 0.010 0.012 0.041 0.049 0.102 0.120 0.004 0.002 0.018
C9 0.021 0.016 0.010 0.008 0.042 0.033 0.103 0.082 -0.004 -0.002 -0.021
C15 0.011 0.010 0.005 0.005 0.021 0.021 0.053 0.052 0.000 0.000 -0.001
C16 0.010 0.014 0.005 0.007 0.021 0.028 0.051 0.070 0.004 0.002 0.020
C17 0.020 0.024 0.010 0.012 0.041 0.049 0.102 0.120 0.004 0.002 0.018
C19 0.021 0.016 0.010 0.008 0.042 0.033 0.103 0.082 -0.004 -0.002 -0.021
C30 0.017 0.021 0.008 0.010 0.035 0.042 0.086 0.104 0.004 0.002 0.018
C31 0.016 0.024 0.008 0.012 0.033 0.049 0.081 0.122 0.008 0.004 0.041
C32 0.010 0.010 0.005 0.005 0.020 0.020 0.050 0.050 0.000 0.000 0.000
C34 0.009 0.008 0.004 0.004 0.018 0.016 0.045 0.040 -0.001 0.000 -0.005
C40 0.017 0.021 0.008 0.010 0.035 0.042 0.086 0.104 0.004 0.002 0.018
C41 0.016 0.024 0.008 0.012 0.033 0.049 0.081 0.122 0.008 0.004 0.041
C42 0.010 0.010 0.005 0.005 0.020 0.020 0.050 0.050 0.000 0.000 0.000
C44 0.009 0.008 0.004 0.004 0.018 0.016 0.045 0.040 -0.001 0.000 -0.005
The results of Table 4 predict that the C9 and C19 sites are the most favored sites face to electrophilic attacks. In the case of
Table 5 gathers the results of local index calculations and dual reactivity descriptors, using MPA population analysis at the computation level B3LYP/6 311G(d,p). It emerges that an electrophilic attack probably occurs on the C9 and C19 atoms, because the dual descriptors take negative values for these latter sites. However, C31 and C41 sites are more favorable to nucleophilic attacks.
As shown in the previous tables, the local indices as well as the dual descriptors derived from the MPA analysis predict a maximum value for the C9 and C19 atoms by promoting them for an
electrophilic attack. Moreover, based on the values of the various indices, we can conclude the nucleophilic attack occurs primarily on carbon C34 and C45.
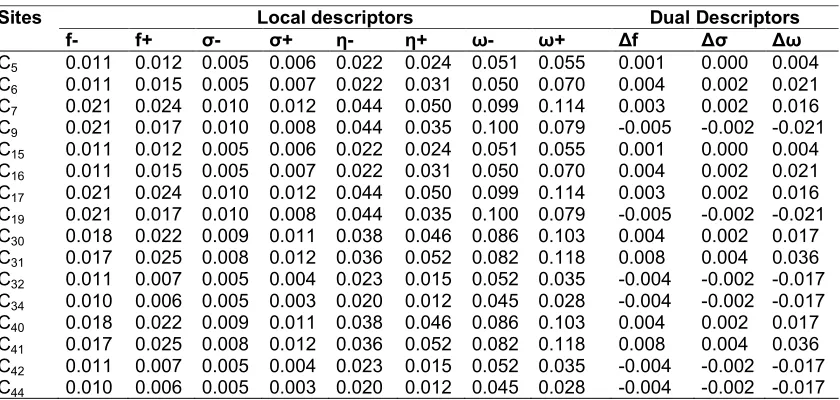
At the end of our study, we can note that the local and dual reactivity descriptors computed using the B3LYP method with the 6-311G (d, p) base suggest that it is the C32 and C42 carbons that are the most favorable sites for electrophilic attacks. This results disagree with those previously obtained with compounds 1 to 4 for which, the C9 and C19 sites are the most privileged for the electrophilic attack.
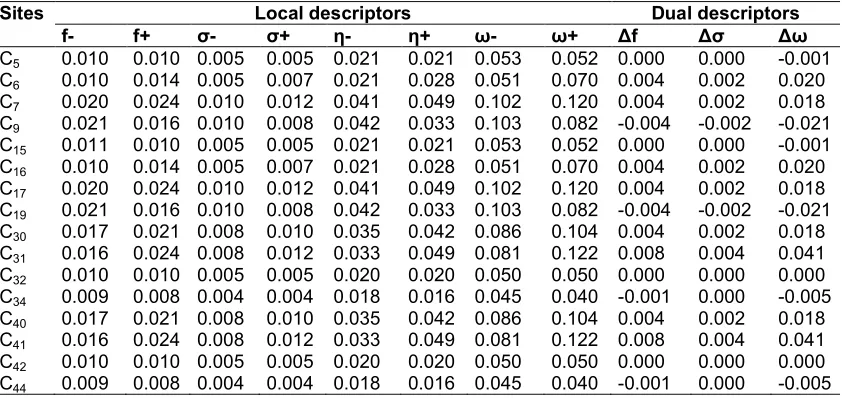
Table 5. Reactivity descriptors of compound 3 computed using Mulliken population analysis (MPA)
Sites Local descriptors Dual Descriptors
f- f+ σ- σ+ η- η+ ω- ω+ Δf Δσ Δω
C5 0.011 0.012 0.005 0.006 0.022 0.024 0.051 0.055 0.001 0.000 0.004
C6 0.011 0.015 0.005 0.007 0.022 0.031 0.050 0.070 0.004 0.002 0.021
C7 0.021 0.024 0.010 0.012 0.044 0.050 0.099 0.114 0.003 0.002 0.016
C9 0.021 0.017 0.010 0.008 0.044 0.035 0.100 0.079 -0.005 -0.002 -0.021
C15 0.011 0.012 0.005 0.006 0.022 0.024 0.051 0.055 0.001 0.000 0.004
C16 0.011 0.015 0.005 0.007 0.022 0.031 0.050 0.070 0.004 0.002 0.021
C17 0.021 0.024 0.010 0.012 0.044 0.050 0.099 0.114 0.003 0.002 0.016
C19 0.021 0.017 0.010 0.008 0.044 0.035 0.100 0.079 -0.005 -0.002 -0.021
C30 0.018 0.022 0.009 0.011 0.038 0.046 0.086 0.103 0.004 0.002 0.017
C31 0.017 0.025 0.008 0.012 0.036 0.052 0.082 0.118 0.008 0.004 0.036
C32 0.011 0.007 0.005 0.004 0.023 0.015 0.052 0.035 -0.004 -0.002 -0.017
C34 0.010 0.006 0.005 0.003 0.020 0.012 0.045 0.028 -0.004 -0.002 -0.017
C40 0.018 0.022 0.009 0.011 0.038 0.046 0.086 0.103 0.004 0.002 0.017
C41 0.017 0.025 0.008 0.012 0.036 0.052 0.082 0.118 0.008 0.004 0.036
C42 0.011 0.007 0.005 0.004 0.023 0.015 0.052 0.035 -0.004 -0.002 -0.017
C44 0.010 0.006 0.005 0.003 0.020 0.012 0.045 0.028 -0.004 -0.002 -0.017
Table 6. Reactivity descriptors of compound 4 computed using Mulliken population analysis (MPA)
Sites Local descriptors Dual descriptors
f- f+ σ- σ+ η- η+ ω- ω+ Δf Δσ Δω
C5 0.007 0.006 0.004 0.003 0.015 0.011 0.042 0.031 -0.002 -0.001 -0.011
C6 0.011 0.008 0.006 0.004 0.022 0.016 0.064 0.045 -0.003 -0.002 -0.018
C7 0.021 0.019 0.011 0.010 0.040 0.038 0.117 0.110 -0.001 -0.001 -0.007
C9 0.041 0.018 0.021 0.009 0.081 0.035 0.236 0.103 -0.023 -0.012 -0.133
C15 0.007 0.006 0.004 0.003 0.014 0.011 0.042 0.031 -0.002 -0.001 -0.011
C16 0.011 0.008 0.006 0.004 0.022 0.016 0.064 0.045 -0.003 -0.002 -0.019
C17 0.021 0.019 0.011 0.010 0.040 0.038 0.117 0.110 -0.001 -0.001 -0.007
C19 0.042 0.018 0.021 0.009 0.081 0.035 0.236 0.103 -0.023 -0.012 -0.133
C30 0.014 0.012 0.007 0.006 0.028 0.023 0.080 0.066 -0.003 -0.001 -0.014
C32 0.009 0.018 0.005 0.009 0.018 0.035 0.052 0.101 0.009 0.004 0.049
C34 0.004 0.035 0.002 0.018 0.007 0.069 0.021 0.201 0.032 0.016 0.180
C35 0.015 0.025 0.008 0.013 0.030 0.049 0.086 0.142 0.010 0.005 0.056
C41 0.014 0.012 0.007 0.006 0.028 0.023 0.080 0.066 -0.003 -0.001 -0.014
C43 0.009 0.018 0.005 0.009 0.018 0.035 0.052 0.101 0.009 0.004 0.049
C45 0.004 0.035 0.002 0.018 0.007 0.069 0.021 0.201 0.032 0.016 0.180
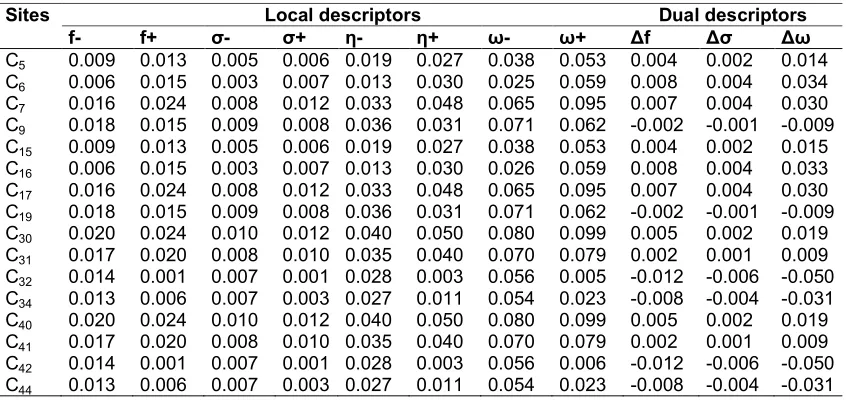
Table 7. Reactivity descriptors of compound 5 computed using Mulliken population analysis (MPA)
Sites Local descriptors Dual descriptors
f- f+ σ- σ+ η- η+ ω- ω+ Δf Δσ Δω
C5 0.009 0.013 0.005 0.006 0.019 0.027 0.038 0.053 0.004 0.002 0.014
C6 0.006 0.015 0.003 0.007 0.013 0.030 0.025 0.059 0.008 0.004 0.034
C7 0.016 0.024 0.008 0.012 0.033 0.048 0.065 0.095 0.007 0.004 0.030
C9 0.018 0.015 0.009 0.008 0.036 0.031 0.071 0.062 -0.002 -0.001 -0.009
C15 0.009 0.013 0.005 0.006 0.019 0.027 0.038 0.053 0.004 0.002 0.015
C16 0.006 0.015 0.003 0.007 0.013 0.030 0.026 0.059 0.008 0.004 0.033
C17 0.016 0.024 0.008 0.012 0.033 0.048 0.065 0.095 0.007 0.004 0.030
C19 0.018 0.015 0.009 0.008 0.036 0.031 0.071 0.062 -0.002 -0.001 -0.009
C30 0.020 0.024 0.010 0.012 0.040 0.050 0.080 0.099 0.005 0.002 0.019
C31 0.017 0.020 0.008 0.010 0.035 0.040 0.070 0.079 0.002 0.001 0.009
C32 0.014 0.001 0.007 0.001 0.028 0.003 0.056 0.005 -0.012 -0.006 -0.050
C34 0.013 0.006 0.007 0.003 0.027 0.011 0.054 0.023 -0.008 -0.004 -0.031
C40 0.020 0.024 0.010 0.012 0.040 0.050 0.080 0.099 0.005 0.002 0.019
C41 0.017 0.020 0.008 0.010 0.035 0.040 0.070 0.079 0.002 0.001 0.009
C42 0.014 0.001 0.007 0.001 0.028 0.003 0.056 0.006 -0.012 -0.006 -0.050
C44 0.013 0.006 0.007 0.003 0.027 0.011 0.054 0.023 -0.008 -0.004 -0.031
Local indices of reactivity ( , and ) as well as the dual descriptors ( ∆ , ∆ and ∆ ), provide the highest value at the C6 and C16 sites. Thus the latter should be the most reactive sites with regard to a nucleophilic attack.
4. CONCLUSION
The maps of electrostatic potentials showed that areas of high electron densities are located around the heteroatoms. The environments of the carbon atoms are globally positive. The global and local indices and the dual reactivity descriptors were calculated. The analysis of the frontier molecular orbitals and the global reactivity indices showed that compound 4 is the most polarizable, has the highest chemical reactivity and the lowest kinetic stability among the studied molecules. In contrast, compound 5 is shown to be the least polarizable with the lowest chemical reactivity and high kinetic stability. Local descriptors obtained by the Mulliken population analysis method indicate that the C9 and C19 atoms for compounds 1 to 4 or C32 and C42 of compound 5 are the preferred sites of electrophilic attack. These interesting results can be used as a precursor for the synthesis of new Schiff bases derived from Dapsone.
COMPETING INTERESTS
Authors have declared that no competing interests exist.
REFERENCES
1. Aide-mémoire N°139, Aout; 2013. 2. Aide-mémoire N°297, Février; 2014. 3. Richard DS, Joanna C. Bull. World Health
Organ. 2002;80:126.
4. Gaüzère BA. La lèpre ou Maladie de Hansen. 2016;1.
5. Mehta PD, Pathak AK. Synthesis,
characterization and in vitro antimicrobial activity of Novel 4,4′-bis[3-chloro-4-aryl-azetidin-2-one-1-yl]diphenyl sulphones. Bulletin of Pharmaceutical Research. 2011;1(3):38-48.
6. Vibhute Yeshwant B, et al. Comparative study of conventional and microwave assisted synthesis of novel schiff bases and their antimicrobial screenings. J. Chem. Pharm. Res. 2010;2(6):234-243.
7. Wadher SJ, Puranik MP, Karande NA,
Yeole PG. Synthesis and biological evaluation of Schiff base of dapsone and their derivative as antimicrobial agents, International Journal of Pharm. Tech. Research. 2009;(1):22-33.
9. N’dri JS, Koné MGR, Kodjo CG, Kablan ALC, Ouattara L, Ouattara O, Ziao N. Combining of DFT and QSAR results to predict the antibacterial activity of a series of azetidinones derived from dapsone as inhibitors of Bacillus Subtilis and
Pseudomonas aeruginosa. SDRP Journal of Computational Chemistry & Molecular Modelling. 2018;2(2):1-9,.
10. Foresman JB, Frisch A. Exploring
chemistry with electronic structure methods, Gaussian Inc., Pittsburgh, USA; 1996.
11. M Kurt, TR. Sertbakan M. Ozduran
Spectrochim. An experimental and
theoretical study of molecular structure and vibrational spectra of 3-and 4-pyridineboronic acid molecules by density functional theory calculations. Acta Part A: Mol. Biomol. Spectrosc. 2008;70:664,. 12. Abdel-Rahman LH, El-Khatib RM, Nassr
LAE, Abu-Dief AM, Ismael M, Seleem AA. Metal based pharmacologically active
agents: Synthesis, structural
characterization, molecular modeling,
CT-DNA binding studies and in vitro
antimicrobial screening of iron(II) bromosalicylidene amino acid chelates, Spectrochimica ActaPart A: Molecular and Biomolecular Spectroscopy. 2014;117: 366–378.
13. Abdel-Rahman LH, Abu-Dief AM, Adam
MSS, Hamdan SK. Some new nano-sized
mononuclear Cu(II) Schiff base
complexes: Design, characterization, molecular modeling and catalytic potentials in benzyl alcohol oxidation. Catal. Lett. 2016;146: 1373–1396.
14. Abdel Rahman LH, Abu-Dief AM, Moustafa
H, Hamdan SK. Ni(II) and Cu(II)
complexes with ONNO asymmetric
tetradentate Schiff base ligand: Synthesis, spectroscopic characterization, theoretical
calculations, DNA interaction and
antimicrobial studies. Appl. Organometal.
Chem. 2017;31:e3555.
15. Ravikumar C, Joe IH, Jayakumar VS, Charge transfer interactions and nonlinear
optical properties of push–pull
chromophore benzaldehyde
phenylhydrazone: A vibrational approach. Chem. Phys. Lett. 2008;460: 552.
16. Parr RG, Yang W. Density-functional theory of the electronic structure of molecules, Annu. Rev. Phys. Chem. 1995;46:701-728.
17. Parr RG, Yang W. Density functional theory of atoms and molecules. Oxford University Press: Oxford, UK; 1989.
18. Fukui K, Yonezawa Y, Shingu H. A
molecular orbital theory of reactivity in aromatic hydrocarbons. J. Chem, Phys. 1952;20:722-725.
19. Parr RG, Yang W. Density functional approach to the frontier-electron theory of chemical reactivity. J. Am. Chem. Soc. 1984;106:4049-4050.
20. Ayers PW, Levy M. Density functional approach to the. Frontier-Electron Theory of Chemical Reactivity. Theor. Chem. Acc. 2000;103(3-4):353-360.
21. Yang Lee W, Parr RG. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B. 1988;37(2):785-789.
22. Becke AD. Density -functional thermo-chemistry. III. The role of exact exchange. J. Chem. Phys. 1993;98(7):5648-5652 23. Gaussian 09, Revision A.02, Frisch MJ, G.
W. Trucks, H. B. Schlegel, G. E. Scuseria, Gaussian, Inc., Wallingford CT; 2009. 24. Kapp J, Remko M, Schleyer PvR. H2XO
and (CH3)2XO Compounds (X= C, Si, Ge, Sn, Pb): Double bonds vs carbene-like structures can the metal compounds exist at all? Journal of the American Chemical Society. 1996;118(24):5745-5751.
25. Johnson BG, Gill PM, Pople JA, The performance of a family of density functional methods. The Journal of Chemical Physics. 1993;98(7):5612-5626.
26. XLSTAT Version 2014.5.03 Copyright
Addinsoft 1995-2014 XLSTAT and
Addinsoft are Registered Trademarks of Addinsoft; 2014.
Available: https://www.xlstat.com
27. Mulliken RS. Criteria for the construction of good self-consistent-field molecular orbital wave functions, and the significance of LCAO-MO population analysis. J. Chem. Phys. 1962;36(12):3428-3439.
28. Koopmans T. Úber die Zuordnung von Wellenfunktiomen und Eigenwerten zu den
einzelnen Elektronen eines Atoms.
Physica. 1934;1:104-113.
29. Yang W, Parr RG, Pucci R. Electron density, Kohn-Sham frontier orbitals, and
Fukui functions. J. Chem. Phys.
1984;81(6):2862-2863.
of the gas-phase basicity of amines. J. Am. Chem. Soc. 1986;108(19):5708–5711. 31. Yang W, Parr RG. Chemistry. Hardness,
softness, and the Fukui function in the electronic theory of metals and catalysis, Proc. Natl. Acad. Sci. USA. 1985;82:6723-6726.
32. Chattaraj PK, Maiti B. Sarkar, Philicity: A unified treatment of chemical reactivity and selectivity. J. Phys. Chem. 2003;107:4973-4975.
33. Fuentealba P, Contreras R. In reviews in modern quantum chemistry: A celebration of the contributions of R. G. Parr; K. D Sen., Ed.; World Scientific: River Edge, NJ, 2002; p 1013. L. R Domingo; M. J. Aurell, P. Perez, R. Contreras. J. Phys. Chem. 2002;106(29):6871-6875.
34. Padmanabhan J, Parthasarathi R, Elango M, Subramanian V, Krishnamoorthy BS, Gutierrez-Oliva S, Toro-Labbe A, et al. Multiphilic descriptor for chemical reactivity and selectivity. J. Phys. Chem. 2007;111: 9130-9138.
35. Morell C, Grand A, Toro-Labbé A. New dual descriptor for chemical reactivity. J. Phys. Chem. 2005;109(1):205-212.
36. Morell C, Grand A, Toro-Labbé A.
Theoretical support for using the Delta f(r). Descriptor. Chem. Phys. Lett. 2006;425: 342–346. (In Press)
37. Abdel-Baset HM, Hanan GE, Mohamed M El-Okr, Medhat A, Ibrahim J. Nanomater Mol Nanotechnol. 2015;4:2.
38. X-Hong L, H-Ling C, R-Zhou Z, X-Zhou Z. Molecular and biomolecular spectroscopy Spectrochimica Acta Part A. 2015;137: 321–327.
39. Woodward RB, Hoffmann R. The
conservation of orbital symmetry. Chemie: Weinhein; Germany; 1970.
40. Gece G. The use of quantum chemical methods in corrosion inhibitor studies. Corros Sci. 2008;50(11):2981-2992.
41. Lewis DFV, Ioannides C, Parke DV.
Interaction of a series of nitriles with the
alcohol-inducible isoform of P450:
Computer analysis of structure-activity relationships. Xenobiotica. 1994;24(5):401-408.
42. Uesugi Y, Mizuno M, Shimojima A,
Takahashi H. Transient resonance raman and ab-initio mo calculation studies of the structures and vibrational assignments of the t1 state and the anion radical of coumarin and its isotopically substituted analogues. J. Phys. Chem. 1997;101(3): 268-274.
43. Sinha L, Prasad O, Narayan V, Shukla S, Raman R. FT-IR spectroscopic analysis and first-order hyperpolarisability of 3-benzoyl-5-chlorouracil by first principles. Molecular Simulation. 2011;37:153-163. _________________________________________________________________________________ © 2018 N’driet al.; This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Peer-review history: