Overview of biological database mapping
services for interoperation between
different ‘omics’ datasets
Shweta S. Chavan,1,2John D. Shaughnessy Jr2and Ricky D. Edmondson2
1University of Arkansas Little Rock (UALR)/University of Arkansas Medical Sciences (UAMS) Joint Bioinformatics Program,
AR 72204, USA
2Myeloma Institute for Research and Therapy, UAMS, Little Rock, AR 72205, USA
*Correspondence to:Tel:þ1 501 454 2764; Fax:þ1 501 686 6442; E-mail: [email protected]
Date received: 17th May 2011
Abstract
Many primary biological databases are dedicated to providing annotation for a specific type of biological molecule such as a clone, transcript, gene or protein, but often with limited cross-references. Therefore, enhanced
mapping is required between these databases to facilitate the correlation of independent experimental datasets. For example, molecular biology experiments conducted on samples (DNA, mRNA or protein) often yield more than one type of ‘omics’ dataset as an object for analysis (eg a sample can have a genomics as well as proteomics expression dataset available for analysis). Thus, in order to map the two datasets, the identifier type from one dataset is required to be linked to another dataset, so preventing loss of critical information in downstream analysis. This identifier mapping can be performed using identifier converter software relevant to the query and target identifier databases. This review presents the publicly available web-based biological database identifier converters, with comparison of their usage, input and output formats, and the types of available query and target database identifier types.
Keywords:omics, genomics, proteomics, identifier mapping, biological database identifier converter
Introduction
Many primary biological databases are dedicated to providing annotation for a specific type of biologi-cal molecule such as a clone, transcript, gene or protein (eg the National Center for Biotechnology
Information [NCBI] Entrez Gene1 database
pro-vides annotation for genes, whereas UniprotKB2,3
provides this for proteins). Other types of second-ary databases provide relevant information about the attributes of these molecules, such as pathway, function(s) or structural information (eg the Kyoto
Encyclopedia of Genes and Genomes [KEGG]4
and Protein Data Bank [PDB].5 Often, these
data-bases provide limited cross-references for
interoperation between databases. Thus, enhanced mapping between these databases is required to facilitate the correlation of independent experimen-tal datasets, which can be provided by Identifier (Id) mapping services.
Id mapping services are tools to connect one type of database Id to the corresponding Id in another database. This mapping includes three types of relationships: one-to-one, one-to-many
and many-to-many. One-to-many and
cell. Also, gene expression datas such as microarray datas are known to have multiple probes targeting a
single transcript and vice versa (eg Affymetrix6
probes, which can be described by many-to-many relationships). Thus, mapping Ids of multiple data-bases to one another facilitates the correlation of different types of ‘omics’ datasets which, in turn, might provide meaningful insights into the biologi-cal processes occurring in a cell.
Several Id mapping services are publicly available (Table 1). The seven Id converters that are discussed in detail in this review were selected to represent the majority of Id converters—as well as major biological databases—used in high-throughput genomics and proteomics datasets. They have many common fea-tures, such as: (i) supporting one-to-many and many-to-many relationships; (ii) providing mapping counts, and the details of the database Ids used as a ‘bridge’ to link to target database Ids; (iii) providing a web-based graphical user interface (GUI) which allows sub-mission of a single Id or a batch search for multiple Ids; and (iv) the output database Ids are hyperlinked to their original database for reference.
These Id converters also differ in a number of ways, such as in the availability of (i) type of query
input; (ii) target output databases; (iii) species; (iv) data sources—and therefore coverage of the genome or proteome of a particular species; (v) ease of use; (vi) database update frequency; (vii) possible conver-sion types (eg protein-to-gene, gene-to-transcripts etc.); (viii) speed of conversion; (ix) a detailed help section or tutorial describing the intended use of the application; and (x) an algorithm for mapping database Ids. In general, these services establish mapping links using existing cross-references or by using sequence alignment information to determine the match. Fewer Id converters have their own published algorithm for establishing the mapping, in addition to using the existing cross-references.
Many of the Id converters provide a web-based intuitive user interface, generally having three com-ponents: input types (ie query databases), output types (ie target databases) and the species under consideration. One such web application is Clone/
Gene ID Converter.7 It provides the option for
several query and target databases for human, mouse and rat species. The output can be custo-mised by selecting from a number of output data-bases which are divided into several logical levels, such as gene, gene clone, protein and functional annotation. Further, detailed references are pro-vided for the resultant output Ids by hyperlinking them to their original data sources. The output can also be obtained in a spreadsheet or text format. A detailed list of input/output databases, availability and other pertinent features can be found in Table 2. A useful piece of information provided on the interface itself includes the specific version that was used as the data source for individual databases. This is of importance, considering the frequent updates of sequence databases and the increasing novel findings about the biological molecules in the respective research areas.
Another similar type of Id converter is ID
mapping hosted by UniProt.3 It supports almost all
organisms, with monthly updates, and maps approxi-mately 90 database sources, including primary
sequence databases and secondary functional/
structural annotation databases. Thus, the input and output database option is divided into Uniprot, other
sequence database, three-dimensional structure,
Table 1. Link to web interface of various Id converters
Mapping services Link
Gene/Clone ID converter http://idconverter. bioinfo.cnio.es/
ID mapping by UniProt http://www.uniprot.org/ ?tab=mapping
MatchMiner http://discover.nci.nih. gov/matchminer/ MatchMinerLookup.jsp
DAVID gene ID conversion tool
http://david.abcc.ncifcrf. gov/conversion.jsp
g:Convert http://biit.cs.ut.ee/ gprofiler/gconvert.cgi
CRONOS http://mips.helmholtz-muenchen.de/genre/ proj/cronos/index.html
bioDBnet:db2db http://biodbnet.abcc. ncifcrf.gov/db/db2db.php
SOFTWARE REVIEW Chavanet al.
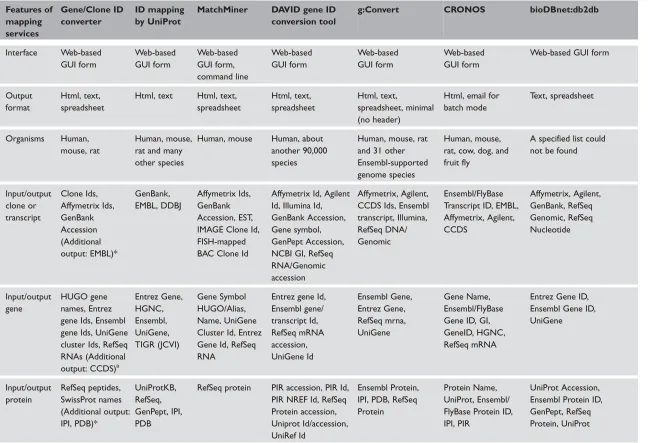
Table 2. Comparison of various Id converters Features of mapping services Gene/Clone ID converter ID mapping by UniProt
MatchMiner DAVID gene ID conversion tool
g:Convert CRONOS bioDBnet:db2db
Interface Web-based GUI form Web-based GUI form Web-based GUI form, command line Web-based GUI form Web-based GUI form Web-based GUI form
Web-based GUI form
Output format
Html, text, spreadsheet
Html, text Html, text, spreadsheet Html, text, spreadsheet Html, text, spreadsheet, minimal (no header)
Html, email for batch mode
Text, spreadsheet
Organisms Human, mouse, rat
Human, mouse, rat and many other species
Human, mouse Human, about another 90,000 species
Human, mouse, rat and 31 other Ensembl-supported genome species
Human, mouse, rat, cow, dog, and fruit fly
A specified list could not be found
Input/output clone or transcript Clone Ids, Affymetrix Ids, GenBank Accession (Additional output: EMBL)* GenBank, EMBL, DDBJ Affymetrix Ids, GenBank Accession, EST, IMAGE Clone Id, FISH-mapped BAC Clone Id
Affymetrix Id, Agilent Id, Illumina Id, GenBank Accession, Gene symbol, GenPept Accession, NCBI GI, RefSeq RNA/Genomic accession
Affymetrix, Agilent, CCDS Ids, Ensembl transcript, Illumina, RefSeq DNA/ Genomic
Ensembl/FlyBase Transcript ID, EMBL, Affymetrix, Agilent, CCDS Affymetrix, Agilent, GenBank, RefSeq Genomic, RefSeq Nucleotide Input/output gene HUGO gene names, Entrez gene Ids, Ensembl gene Ids, UniGene cluster Ids, RefSeq RNAs (Additional output: CCDS)a
Entrez Gene, HGNC, Ensembl, UniGene, TIGR (JCVI) Gene Symbol HUGO/Alias, Name, UniGene Cluster Id, Entrez Gene Id, RefSeq RNA
Entrez gene Id, Ensembl gene/ transcript Id, RefSeq mRNA accession, UniGene Id Ensembl Gene, Entrez Gene, RefSeq mrna, UniGene Gene Name, Ensembl/FlyBase Gene ID, GI, GeneID, HGNC, RefSeq mRNA
Entrez Gene ID, Ensembl Gene ID, UniGene Input/output protein RefSeq peptides, SwissProt names (Additional output: IPI, PDB)* UniProtKB, RefSeq, GenPept, IPI, PDB
RefSeq protein PIR accession, PIR Id, PIR NREF Id, RefSeq Protein accession, Uniprot Id/accession, UniRef Id
Ensembl Protein, IPI, PDB, RefSeq Protein
Protein Name, UniProt, Ensembl/ FlyBase Protein ID, IPI, PIR
protein– protein interaction, protein family/group, two-dimensional gel, genome annotation, organism specific gene database, phylogenomics, enzyme and pathway and gene expression, and other database types which are listed in Table 2. The output is pro-vided in the form of a tab-delimited table indicating the Ids in the query that could be mapped to those in the target databases, along with a list of unique target database Ids and a list of those Ids that could not be mapped. Id mapping by UniProt also provides an application programming interface (API) for pro-grammatic access, as well as file transfer protocol (FTP) downloads if the user wishes to have a local Id
mapping service for large datasets (.100,000 Ids).
MatchMiner8 is tool that provides a clean
inter-face with an interesting BatchMerge option, along
with Interactive Lookup and Batch Lookup.
Interactive Lookup and Batch Lookup can be used for generic Id conversion for single and multiple query input, respectively. The Batch Merge option is intended to be used to merge an input of two different query database Id lists into a single list, by determining which of the Ids from the two lists refers to the same gene or biological entity. In the Id conversion html output, hyperlinks are provided from each output Id to the original database for
some (eg Entrez Gene, UniGene9), but not all,
databases (eg Affymetrix). Also, MatchMiner
follows a hierarchy of source reliability while searching for an Id and specifies the source database in the output; for example, if the input Id is a
GenBank10 Accession, then the algorithm first
searches for the Id from University of California
Santa Cruz (UCSC)11-known genes. If this is not
found, only then it does search through UniGene and then UCSC expressed sequence tags (ESTs). Details of hierarchy of source reliability for all source databases can be found in the original article on
MatchMiner.8 Another unique feature of
MatchMiner is that it provides a command line interface option for querying, which can be useful in cases where MatchMiner is to be integrated as a part of a pipeline or as a filtering step in a workflow. This feature requires certain system requirements; details can be found at http://discover.nci.nih.gov/
matchminer/command.jsp. Thus, MatchMiner
T able 2. C o n tinued F e a tur es of mapping ser vices Gene/Clone ID con v erter ID mapping b y UniPr ot Ma tchMiner D A VID g ene ID con v ersion tool g:C on v ert CR ONOS bioDBnet:db2db
Input/output other informa
tion (Additional output: PubMed, GO , KEGG, R e actome, Chr omosomal loca tions fr om Ensembl, UCSC
Genome Bro
wser , OMIM) a SGD , GeneRif, NCBI T axon, and others Cytogenetic loca tion: UCSC (Additional output: PubMed, GO , KEGG, R e actome, Chr om osomal loca tions fr om Ensembl, UCSC Genome Br o wser , OMIM)* “Not sur e” type also accepted, and man y other secondar y da tabase ident ifiers also supported UCSC , PubMed, GO and man y other secondar y d atabases dbSNP , UniSTS, MGI, orfnames, MIM, MORBID , CDD GO , InterPr o , Biocarta, KEGG, dbSNP , H-In vita tional (H-In v), HomoloGene, MGC , MIM, UniSTS, T axon , and other secondar y d atab ases aAll input Id types ar e potential output Id types as w ell (eg): as in Id mapping by UniPr ot. In some cases, ho w e ve r, ther e ar e additional output Id types av ailable to choose fr om which ar e not available as input Id using tha t particular conv erter . Such output Id types ar e mentioned in par entheses and ar e indica ted as ‘additional output’ (eg) as in Clone/Gene Id conv erter , GUI gr aphical user interfa ce.
SOFTWARE REVIEW Chavanet al.
provides certain unique features that can be useful for specialised Id conversion needs.
Most of the Id converters use the available data sources to create mapping; however, the DAVID
gene ID conversion tool12 – 14 uses its own
knowl-edge base, which is based on the DAVID gene
concept,13 in addition to the primary Entrez- and
Uniprot-based mapping. The data source used by this tool includes 20 main gene/protein Id types, in addition to other secondary Id types. It also has the capability to handle a mixture of Id types in case of ‘unsure’ input Id type. The output yields summary statistics for conversion, including Id count, presence in DAVID database and conversion status as success-ful or otherwise, with possible choices for ambigu-ities, such as when the input Id may not belong to the database as specified by the user but may exist as
an Id in another database. The DAVID
Knowledgebase is available for download as well.
bioDBnet15 provides a converter ‘db2db’ which
has wide coverage of databases, including 153 data-base Id types including genes, proteins, pathways and other biological concepts as their data source. It also provides other menu options for Id conver-sion, such as ‘dbFind’, when the input Id type is unknown, ‘dbWalk’, where the user can direct the type of conversion and the intermediate databases to ‘walk’ through, ‘dbReport’, which provides an all-inclusive search, one Id to all other available Ids/annotations available. Thus, bioDBnet provides flexible interface options and, importantly, is updated weekly. Likewise, g:Convert, which is a part
of g:Profiler,16 provides mappings which are mainly
based on the Ensembl database,17 created through a
three-level index of gene, transcript and protein Ensembl Ids. By contrast, the cross-reference
naviga-tion server (CRONOS)18 provides mappings which
are based on primary resources such as Uniprot,
RefSeq19and Ensembl. These mappings are validated
by eliminating ambiguous gene names, which provide an all-inclusive search of one Id to all other available Ids/annotations available.
Conclusion
This review is by no means comprehensive, but is intended to be representative of the currently available
Id converters. Thus, there are several other Id conver-ters that are part of other integrative analysis systems which are not reviewed here but might be of interest
to researchers—such as Babelomics,20 BioMart,21 ID
Converter System,22 BridgeDB etc.23 Many of the
users provide their feedback after using these tools at internet forums (eg http://biostar.stackexchange.com/ questions/22/gene-id-conversion-tool). Comparisons are made using a test set of Ids to test the performance of different Id converters (eg http://www.scribd.com/ doc/18966500/Id-Converters-Test) that might aid in the selection of an appropriate Id converter. Such comparative analysis is not presented in this review, as the intended use of each of the Id converters is differ-ent and each has its own unique features which may not be measured by direct comparison. It is, however, recommended that one should base the choice of an Id converter application on the researcher’s conversion needs; for example, the availability of the required input and output Id type, acceptable mapping algor-ithm and database update frequency, which are described in this review and summarised in Table 2, as well as other factors that might be of interest for the biological experiment being conducted.
Acknowledgments
The authors thank the Nancy and Stephen Grand Philanthropic Foundation, Myeloma Institute for Research and Therapy, UAMS, Multiple Myeloma Research Foundation and the IDeA Networks of Biomedical Research Excellence (INBRE) Program of the National Center for Research Resources.
References
1. Maglott, D.R., Ostell, J., Pruitt, K.D. and Tatusova, T.A. (2005), ‘Entrez Gene: Gene-centered information at NCBI’,Nucleic Acids Res. Vol. 33, pp. 54 – 58.
2. Apweiler, R., Bairoch, A., Wu, C.H., Barker, W.C. et al. (2004), ‘UniProt: The Universal Protein knowledgebase’, Nucleic Acids Res. Vol. 32, pp. 115 – 119.
3. Bairoch, A., Apweiler, R., Wu, C.H., Barker, W.C.et al. (2007), ‘The Universal Protein Resource (UniProt)’, Nucleic Acids Res. Vol. 33, pp. 154 – 159.
4. Kanehisa, M. and Goto, S. (2000), ‘KEGG: Kyoto Encyclopedia of Genes and Genomes’,Nucleic Acids Res.Vol. 28, pp. 27 – 30.
5. Sussman, J.L., Lin, D., Jiang, J., Manning, N.O.et al. (1998), ‘Protein Data Bank (PDB): Database of three-dimensional structural information of biological macromolecules’,Acta Crystallogr.Vol. 54, pp. 1078 – 1084. 6. Liu, G., Loraine, A.E., Shigeta, R., Cline, M.S.et al. (2003), ‘NetAffx:
7. Alibe´s, A., Yankilevich, P., Can˜ada, A. and Dı´az-Uriarte, R. (2007), ‘IDconverter and IDClight: Conversion and annotation of gene and protein IDs’,BMC BioinformaticsVol. 8, p. 9.
8. Bussey, K.J., Kane, D., Narasimhan, S., Nishizuka, S. et al. (2003), ‘MatchMiner: A tool for batch navigation among gene and gene product identifiers’,Genome Biol.Vol. 4, p. R27.
9. Pontius, J.U., Wagner, L. and Schuler, G.D. (2002), ‘Unigene: A unified view of the transcriptome’, in: McEntyre, J. and Ostell, J. (eds),The
NCBI Handbook, NCBI, Bethesda, MD, pp. 277 – 288.
10. Benson, D.A., Karsch-Mizrachi, I., Lipman, D.J., Ostell, J.et al. (2002), ‘GenBank’,Nucleic Acids Res. Vol. 30, pp. 17 – 20.
11. Kent, W.J., Sugnet, C.W., Furey, T.S., Roskin, K.M.et al. (2002), ‘The Human Genome Browser at UCSC’,ScienceVol. 12, pp. 996 – 1006. 12. Huang, D.W., Sherman, B.T. and Lempicki, R.A. (2009),
‘Bioinformatics enrichment tools: Paths toward the comprehensive func-tional analysis of large gene lists’,Nucleic Acids Res.Vol. 37, pp. 1 – 13. 13. Sherman, B.T., Huang, D.W., Tan, Q., Guo, Y.et al. (2007), ‘DAVID
Knowledgebase: A gene-centered database integrating heterogeneous gene annotation resources to facilitate high-throughput gene functional analysis’,BMC BioinformaticsVol. 8, p. 11.
14. Huang, D.W., Sherman, B.T. and Lempicki, R.A. (2009), ‘Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources’,Nat. Protoc.Vol. 4, pp. 44 – 47.
15. Mudunuri, U., Che, A., Yi, M. and Stephens, R.M.. (2009), ‘bioDBnet: The biological database network’, Comput. Appl. Biosci. Vol. 25, pp. 555 – 556.
16. Reimand, J., Kull, M., Peterson, H., Hansen, J. et al. (2007), ‘g: Profiler — A web-based toolset for functional profiling of gene lists from large-scale experiments’,Nucleic Acids Res.Vol. 35, pp. 193 – 200. 17. Waegele, B., Dunger-Kaltenbach, I., Fobo, G., Montrone, C. et al.
(2009), ‘CRONOS: The cross-reference navigation server’, Comput.
Appl. Biosci.Vol. 25, pp. 141 – 143.
18. Hubbard, T., Barker, D., Birney, E., Cameron, G. et al. (2002), ‘The Ensembl genome database project’,Nucleic Acids Res. Vol. 30, pp. 38 – 41. 19. Pruitt, K.D., Tatusova, T.A. and Maglott, D.R. (2005), ‘NCBI Reference Sequence (RefSeq): A curated non-redundant sequence database of genomes, transcripts and proteins’, Nucleic Acids Res. Vol. 33, pp. 501 – 504.
20. Al-Shahrour, F., Minguez, P., Ta´rraga, J., Montaner, D. et al. (2006), ‘BABELOMICS: A systems biology perspective in the functional annota-tion of genome-scale experiments’, Nucleic Acids Res. Vol. 34, pp. 472 – 476.
21. Smedley, D., Haider, S., Ballester, B., Holland, R. et al. (2009), ‘BioMart — Biological queries made easy’, BMC Genomics Vol. 10, p. 12.
22. Imanishi, T. and Nakaoka, H. (2009), ‘Hyperlink Management System and ID Converter System: Enabling maintenance-free hyperlinks among major biological databases’, Nucleic Acids Res. Vol. 37, pp. W17 – W22.
23. Van Lersel, M.P., Pico, A.R., Kelder, T., Gao, J. et al. (2010), ‘The BridgeDb framework: Standardized access to gene, protein and metab-olite identifier mapping services’,BMC BioinformaticsVol. 11, p. 7.
SOFTWARE REVIEW Chavanet al.