Copyright 0 1997 by the Genetics Society of America
Enhanced Deletion Formation by Aberrant DNA Replication
in
Escherichia coli
Catherine
J. Saveson
and
Susan T.
Lovett
Department of Biology and Rosenstiel Basic Medical Sciences Research Center, Brandeis University,
Waltham, Massachusetts 02254-91 10
Manuscript received September 26, 1996 Accepted for publication February 21, 1997
ABSTRACT
Repeated genes and sequences are prone to genetic rearrangements including deletions. We have
investigated deletion formation in Eschm'chia coli strains mutant for various replication functions. Deletion
was selected between 787 base pair tandem repeats carried either on a ColElderived plasmid or on the
E. coli chromosome. Only mutations in functions associated with DNA Polymerase I11 elevated deletion
rates in our assays. Especially large increases were observed in strains mutant in dnaQ the E editing
subunit of Pol 111, and dnuB, the replication fork helicase. Mutations in several other functions also
altered deletion formation: the a polymerase ( d n a , the y clamp loader complex (holC, dnaX), and
the
p clamp
(dnaN) subunits of Pol I11 and the primosomal proteins, dnaCand pui. Aberrant replicationstimulated deletions through several pathways. Whereas the elevation in dnaB strains was mostly recA-
and Zeddependent, that in dnaQstrains was mostly recA- and Zed-independent. Deletion product analysis
suggested that slipped mispairing, producing monomeric replicon products, may be preferentially in-
creased in a dnaQ mutant and sister-strand exchange, producing dimeric replicon products, may be
elevated in dnaE mutants. We conclude that aberrant Polymerase I11 replication can stimulate deletion events through several mechanisms of deletion and via both recA-dependent and independent pathways.
T
ANDEMLY repeated DNA sequences, common inthe genomes of many organisms, are vulnerable to deletions and amplifications. Such rearrangements may occur between repeated genes, kilobases in length, or between short segments, of as little as a few nucleo- tides in length. These tandem repeat rearrangements are common sources of genetic mutation and human
genetic disease (MEUTH 1989; KRAWCZAK and COOPER
1991; Hu and WORTON 1992; NELSON 1993).
Genetic analysis of repeated sequence rearrange-
ments in Escherichia coli has revealed that multiple mech-
anisms contribute to the process. In particular, we have
previously investigated the genetic dependence of spon-
taneous deletion events between 787 base pair (bp)
repeated sequences within the tetA gene of E. coli (Fig-
ure 1 ) (LOVETT et al. 1993). The majority of recovered deletions occur by a homologous recombination path- way requiring the RecA strand transfer protein. How-
ever, a substantial proportion of these events (30%)
occur independently of RecA.
Both RecA-independent and -dependent deletion
formation occurs between homologous target se-
quences. However, there is a minimal homology re-
quirement for the Red-dependent pathway and no
such requirement for the Red-independent pathway. The RecA-dependent mechanism does not appear to
function on homologies less than -200 bp in length
(DIANOV et al. 1991; WIN et al. 1991; BI and LIU 1994).
Corresponding author: Susan T. Lovett, Rosenstiel Basic Medical Sci-
ences Research Center MSO29, Brandeis University, Waltham, MA
0225491 10. E-mail: [email protected]
Genetics 146: 457-470 (June, 1997)
The RecA-independent mechanism can mediate dele- tion between homologies of several nucleotides, al- though the efficiency of deletion is improved dramati-
cally with increased homology (ALBERTINI et al. 1982;
DIANOV et al. 1991; WIN et al. 1991; PIERCE et al. 1991;
BI and LIU 1994).
DNA replication may play a role in both mechanisms
of deletion formation. The events that may trigger
RecA-dependent homologous recombination between
tandem repeats are not known but could include a
blocked replication fork. Our genetic studies impli- cated the RecF, RecO and RecR homologous recombi-
nation proteins of E. coli in the RecAdependent dele-
tion pathway
LOVE^
et d . 1993; M. Bzymek and S. T.Lovett, unpublished results). These functions are also
required for recombinational repair of DNA lesions
that block replication in the cell (TSENG et al. 1994).
Therefore, some tandem rearrangements could result from recombinational gap-filling reactions in a blocked
replication fork as depicted in Figure 2. Unequal cross-
ing-over between sister chromosomes or recombination between the two repeats on the same chromosome dur-
ing the course of this repair may lead to deletion of
one of the repeated segments. Alternatively, as shown
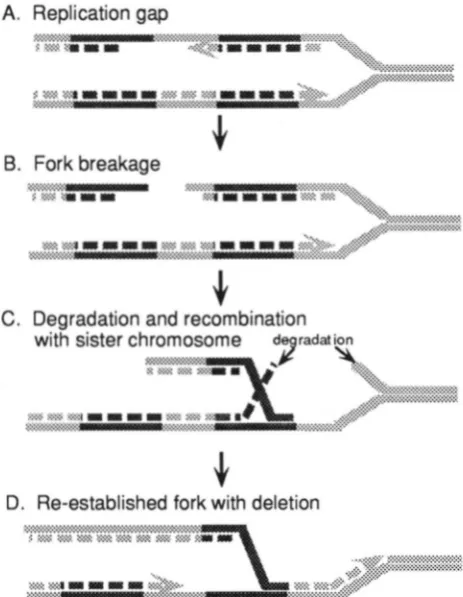
in Figure 3, a stalled replication fork may be broken;
recombination of the broken chromosome with its sis- ter to restore the fork could result in deletion of tandem
repeats (ASAI et al. 1994; KUZMINOV 1995a,b).
458 C. J. Saveson and S. T. Lovett
fetA+
.:.:.:.:.:.:.:.:.:
:.:.:.:.:.:.;.:.:. .:+:+:.:.:.: :.:.:.:.:.:.:.:.
tetAdu
EmRV Nrul:.:.:.:.:.:.:.: ... ...
... ... :::::::::I
FIGLIRE 1.-Diagram of the k t A locus and the 787-bp dupli- cation used in these experiments. Duplication of the segment from the EcoRV to the NmI sites of the gene disrupts the t d A
gene. Deletion of one copy of the tandem repeat restores
the IdA' gene structure and can be selected by tetracycline-
resistance.
cative mechanism for the RecA-independent deletion of both short (TRINH and SINDEN 1991) and long (LO-
VETT and FESCHENKO 1996) tandem repeats. Exposure
of singlestrand DNA during the replication process may provide an opportunity for tandem sequence ho- mologies to interact. A simple slipmispairing model (Figure 4A) proposes that slipped realignment between the nascent strand and its template leads to deletions or amplifications (STREISINCER et al. 1967; ALBERTINI et al. 1982). Additional models propose that deletions may arise by strand misalignment within a replication fork concomitant with sisterchromosome exchange (Figure 4B) (LOVEIT et al. 1993; BI and LIU 1996). The location of these slipped mispairing events within closely juxta- posed and singlestranded regions of the replication fork would explain why a strand transfer protein such as R e d is not needed for such reactions.
Because DNA replication is implicated in both RecA- dependent and -independent deletion mechanisms, we have investigated the genetic relationship between DNA replication components and deletion formation. There are three DNA polymerases in E. coli: Polymerase I11 bears the major responsibility for chromosomal replica- tion, Polymerases I and I1 are both believed to be in- volved in repair pathways. Polymerase I is required for some plasmid replication initiation and may also play a role in Okazaki fragment joining (KORNBERG and BAKER 1992).
Polymerase I11 itself is a complex enzyme with multi- ple subunits encoded by different genes (BAKER and WICKNER 1992; MARIANS 1992; KELMAN and O'DONNELL
1995; MARIANS 1996). The polymerase activity is pro- vided by the a subunit, encoded by dnaE.
Its
proofread- ing e subunit, possessing exonuclease activity, is en- coded by dnaQ. The function of the final subunit of the core polymerase, 8, encoded by hoE, is apparently nonessential (STUDWELL-VAUGHAN and O'DONNELL 1993; SLATER et al. 1994). It is believed that both leading and lagging strands are coordinately synthesized by an asymmetric polymerase dimer (JOHANSON and MCHENRY 1984). The T subunit, encoded by dnuX, facil- itates the interaction between the two core polymerase molecules (MCHENRY 1982; STUDWELL-VAUGHAN andC.
D.
E.
Branch migration
J/
J.
Resynthesis
*rn??%?*?A?Y*?* 3 :;I
:=
K. ~ :..,. .,,,,, I ! R x * ~ : ? ~ w $ $ ~ ? ? x < x x < > ~ I ~ ~<;:=
~ ~ .... :.:.. ..:s., ~ : * ~ .::::., .:+ ... ... n.. 6:: .... .... .... .... ...
.... ... ... ....
... ... ...
... ....
..:.:.>. ... x&
+??
.... ...
.... ... >:.:+>:.:.:.:.:.:.>>:.:.>:.:.:.
... ... ...
<*+-
... ... < ...
... ....
... ... ... ...
... .:.:..
...
.:.:.. .:.:., .*.:.. &.
-< >.*.Mm$$: fi:::..<+w >y* a &% w ->
* *
k.& e g< s:; .:$;*??x%K&Y*;
ss:*...Junction resolution
c
...
FIGURE 2.-Recombinational gapfilling (post-replication) repair. (A) DNA synthesis cannot proceed past a replication- blocking lesion, giving rise to a daughterstrand gap. (B) The gap can be filled by recombination with the sister chromo- some. (C) Branch migration of the Holliday junction can bypass the lesion and cause the incomplete nascent strand to
switch templates. (D) Replication is completed on the sister template. (E) The Holliday junction is resolved. This particu- lar resolution produces a cross-over between sister chromo- somes.
O'DONNELL 1991). The subunit, encoded by dnaN, is a processivity factor that forms a clamp around the DNA to anchor the polymerase (STUKENBERG et al. 1991). This clamp is loaded by a polymerase-associated complex (O'DONNELL 1987), which includes the y
(also encoded by dnaX), 6 (holA),
6'
(holB),x
(holCj,and t,b ( h o D ) proteins.
Hyperdeletion in Replication Mutants 459
A.
B.
C.
J,
JI
Degradation and recombination with sister chromosome dqradatQn
J.
FIGURE 3.-Replication fork collapse and repair (Kuzminov
1995b). (A) Inhibition of polymerization stalls the replication fork. (B) Nuclease attack of single-strand DNA breaks the fork. (C) The broken end invades i t s sister chromosome. Dur- ing this step unequal recombination can occur at a repeated
sequence (dark hatching). (D) The fork is reestablished with
a deletion on one chromosome.
association is mediated by interactions with the dnaB protein, also the replication fork helicase (TOUGU et al.
1994). The primase, like the
P
clamp, is required at the start of each Okazaki fragment, placing a potential greater burden on these functions for lagging strand synthesis. Other proteins that are required for primo- some assembly and may also be components of the pri- mosome include the p i A , @‘B, pzC, dnaC and dnaT gene products(ARAI
and KORNBERG 1981). An alterna- tive primosome assembly occurs at the E. coli origin of replication, using the d n d protein in addition to dnaB and dnaC (FUNNELL et al. 1987; M A S N et al. 1990).In this work, we have investigated whether aberrant replication promotes deletions at repeated sequences. We constructed a series of isogenic strains containing mutations that impair selected components of the repli- cation machinery. Because there are three DNA poly- merases in E. coli, we have tested the genetic depen- dence of deletion on each polymerase gene as well as
other functions associated with DNA replication. E. coli mutant strains were surveyed for effects on deletion formation using both plasmid and chromosomal dele- tion assays. To gain insight into the mechanisms of dele- tion during aberrant replication, we also analyzed the
deletion products from selected strains. We present evi- dence that aberrant or incomplete replication via DNA polymerase I11 stimulates deletion at tandem repeats via several mechanisms.
MATERIALS AND METHODS
Bacterial strains, media and antibiotics: Strains were grown
on LB media: 1 % Bacto-tryptone, 0.5% yeast extract, 0.5%
sodium chloride and 0.005% thymine, and, for plates, 1.5%
agar (WIl.LETTS et dl. 1969). Growth temperatures are indi-
cated in the text. Minimal complete media consisted of 56/
2 salts (WILLETTS et al. 1969), 0.2% glucose, 1 pg/ml thiamine,
50 pg/ml each of required amino acids and, for plates, 2% agar. LCG media for preparation of P1 lysates and transduc- tions consisted of LB media supplemented with 1% glucose
and 2 mM calcium chloride, and, for plates, 1 % agar. A 0.7%
agar concentration was used for LB and LCG top agar. Media
were supplemented with antibiotics: kanamycin ( K m ) at 60
pg/ml, tetracycline (Tc) at 8-15 pg/ml, chloramphenicol
(Cm) at 15 pg/ml and ampicillin (Ap) at 100 pg/ml (for
plasmid resistance genes) and 30 pg/ml (for chromosomal genes).
Isogenic strains used for deletion assays were constructed
by P1 transduction and are described in Table 1 . These strains
include derivatives of AB1 157 for plasmid deletion assays (A)
and STL695 for chromosomal deletion assays (B). P1 u i r A
phage lysates and transduction were performed as described (MILLER 1992). For many strains, a TnlQkan element linked
to the mutation of interest was used for selection in strain
constructions. In these cases, control strains were constructed
carrying only the Tn IOkun element to ensure that this marker
did not contribute to the deletion phenotype. Deletion assays confirmed that deletion rates in these control strains were within 50% of the wild-type value (data not shown). Mutants in dnuQ were assayed for mutator phenotype on rifampin plates (at 100 pg/ml).
Deletion assays: Deletion formation was measured using
plasmid pSTL55 (LOVE= et al. 1993). This plasmid is a deriva-
tive of pBR322, conferring ampicillin resistance, and contains
a tandem repeat within the tetA gene (Figure 1). The dupli-
cated region spans 787 bp between the EcoRV and NruI sites
of tetA. Plasmid DNA was introduced into strains via electro-
poration (DOWER et ul. 1988). Chromosomal deletion fre-
quencies were measured in strains carrying an insertion of
bla and tetAdup787 from pSTL55 on the E. coli chromosome
at lacZ (LOVE= et al. 1993). Deletion of one tandem repeat
results in recovery of a functional tetA gene and confers resis-
tance to tetracycline.
Strains containing either the chromosomal or plasmid con-
struct were grown at their permissive temperatures on LB plates containing ampicillin. Individual colonies were then picked whole, and grown in LB+Ap broth for 2 hr. The cul-
tures were diluted in 56/2 buffer and the number of Ap‘ and
Tc‘ cells in the population was determined by plating cell dilutions on LB+Ap and LB+Ap+Tc media. Because of poor
growth in rich medium, polA and
pnA
mutant strains weregrown on minimal-complete agar plates and in minimal-liquid media with the appropriate antibiotics. Deletion assays were performed for 2 1 0 independent cultures in parallel with the wild-type control strain. Deletion rates were calculated by the
method of the median (Lw and COULSON 1949) for the en-
tire set of assays using the formula: deletion rate = M / N ,
where M is the calculated number of deletion events and N
is the final average number of Ap‘ cells in the 1-ml cultures. Mis solved by interpolation from experimental determination
460 C. J. Saveson and S. T. Lovett
A. Simple slipped mispairing B. Sister-strand slipped mispairing
f Nascent strand
<Template strand
wk::~s7~::;*;;~x ... ..". ...
:.:.:.:. '.' 1 II ...~.:.~.~.:.:.~.:.:~.. ... ... ...
E?%dR???%?
...????>% ... ".::.:.A .,.!? ... ... ... ... ...:*
x.:.:. ... ...
*:<
*
... ..:.&$:-.:.:.x;:.:.:.:-.:~<~<~:~:~~: :.:,.:.:.> ... .... :.:.:..
Sister strands anneal
+
Slipped alignment .., >,>..:.:.:.>:<.:.:.: ... W? VS
...
w.s-<:- ... ... ...
I
*
Monomer deletion product
+
+
Dimer deletion product
FIGURE 4.-Slipped realignment mechanisms for deletion formation. (A) Simple slippage. Displacement and realignment of
a nascent strand with its template can delete one copy of a tandem repeat. On a circular chromosome, such deletion products should be replicon monomers. (B) Sister-strand slipped mispairing. Nascent strands are displaced and mispair with each other, causing a deletion to be formed. [Although we have illustrated leading strand synthesis impeded relative to the lagging strand,
the ends of the nascent strands may be created by nucleolytic cleavage rather than representing the point at which DNA synthesis
has stopped. See LOVEIT et al. (1993) for further discussion of this model.] With pairing of the parental strands, a recombinational
intermediate is produced. Resolution o r replication through the Holliday junction intermediate causes sisterchromosome
exchange. Crossing-over between circular sister chromosomes produces a dimeric deletion product. Both mechanisms are
presumed to operate independently of RecA.
cultures, by the formula r,, = M( 1.24
+
InM).
The observednumbers of Tc' cells for a given data set were ranked and a 95% confidence limit for the median was established using
Table A-25a of DIXON and MASSEV (1969); this was converted
to a 95% confidence interval for deletion rates as suggested
by Wierdl et al. (1996) using the formula above.
Analysis of plasmid deletion products T e d ' plasmid dele- tion products were examined by electrophoresis to determine
monomer and dimer product distribution. These deleted
products were readily distinguishable from each other and from the parental plasmid. Each product examined was iso-
lated from an independent culture. Dimeric plasmid DNA was
never detected from nondeleted Apresistant (Tc-sensitive) colonies, confirming that dimerization occurred concurrent
with deletion formation. Plasmid DNA was prepared from
each culture by alkaline lysis (SAMBROOK et al. 1989) or by
phenolchloroform extraction and subjected to agarose gel
electrophoresis. Phenolchloroform DNA extraction was per- formed as follows: single colonies were randomly selected and
suspended in 25 pl of TE (10 mM Tris-HCI, pH 8.0, 1 mM
EDTA). For slow-growing strains, individual colonies were
streaked to LB+Tc and incubated overnight, producing a
larger streak, which was then suspended in 25 p1 of TE. This
suspension was extracted with 25 pl of phenolchloroform-
isoamyl alcohol (25:24:1 by volume), mixed vigorously for 1 min and cleared by microcentrifugation for 2 min. The entire
supernatant was then subjected to agarose gel electrophoresis.
RESULTS
Effects of replication mutations on deletion forma-
tion: We constructed a series of isogenic strains in the
AB1157 strain background that carry various replication mutations (Table 1). Deletion of tandem direct repeats was measured using
both
a plasmid-based and chrome somal assay. Plasmid pSTL55 (LOVEIT et aZ. 1993), con- taining a 787-bp internal tandem repeat disrupting thetea
gene on plasmid pBR322, was introduced into each strain. Precise deletion of one repeat restores a functionaltea
gene and tetracyline resistance to the cell. The c h r e mosomal assay used the identical 787-bp ,!&A duplication construct integrated into the chromosomallac
gene (Lo-VEIT et al. 1993). In the case of
pnA
and pol4 mutants,deletion rates could be measured only for the chrome somal construct because of poor plasmid maintenance. Deletion rates were calculated from the number of cells in the population that acquired tetracycline resistance (as
described in MATERIAIS AND METHODS).
Hyperdeletion in Replication Mutants 46 1
1994) on rich medium, these strains were grown and
assayed on minimal medium at
37”
(Table2B).
Strainswith temperature-sensitive lethal phenotypes (Table
2,
C and D) were measured at their permissive tempera-
ture for growth, either 25 or 30”. In each case, we show absolute deletion rates for the mutant strain and the rate relative to the wild-type control strain under the same growth conditions (Table 2). For the wild-type strains, the various growth conditions had little effect on the efficiency of deletion formation.
We found that many replication mutants exhibited higher deletion rates in both plasmid and chromosomal
deletion assays (Table
2).
In particular, some mutantswith defects in DNA polymerase I11 core enzyme exhib-
ited high deletion rates. This includes a sevenfold eleva-
tion of plasmid deletion rates seen for the temperature- sensitive dnaE486 (poZC486) mutant, which is defective
in the a polymerase subunit itself. However, a minor
twofold effect on chromosomal deletion rates was seen. Deletion assays of strains carrying the antimutator allele
of dnaE, dnaE925, were normal. Mutants carrying
dnaQ49, affecting the 3’ to 5‘ editing exonuclease E
subunit, exhibited dramatic increases in deletion for- mation for both assays, with ninefold and 100-fold stim- ulation of plasmid and chromosomal deletion rates, re-
spectively. Likewise, another allele of dnuQ, mutD5,
stimulated deletion rates threefold in the plasmid assay and 600-fold in the chromosomal assay. Null mutants in hoE, the third 8 subunit of the polymerase core enzyme complex, were unaffected for deletion formation in ei- ther assay. In contrast to these mutations affecting DNA polymerase 111, deletions of the genes for polymerase I
(poZA) and polymerase I1 (poZB) had no influence on deletion rates.
Mutants in the P-processivity “clamp” associated with polymerase 111, dnaN, gave seemingly contradictory re- sults in the two assays: 14fold higher deletion rates in the plasmid assay and eightfold lowered deletion rates
in the chromosomal assay as compared to wild-type
strains. This latter result is of special interest because it implicates replication as an essential component of deletion formation and suggests that when the polymer-
ase clamp is impaired, formation or recovery of deletion
events on the E, coli chromosome is reduced. The dis-
crepancy between the two assays will be addressed in
the DISCUSSION below.
Other mutants affecting the “clamp loader” (the y
complex of the DNA polymerase I11 holoenzyme) were analyzed. Deletion formation in temperature-sensitive
mutants of the dnaXgene, which encodes both T and
6 subunits of polymerase 111, was at normal levels for the
plasmid assay but 30-fold reduced in the chromosomal assay, reminiscent of the dnaN results above. A null
mutant in hoZC, encoding the
x
subunit of the y com-plex, exhibited a fivefold elevated deletion rate in the plasmid assay and a 16-fold elevated rate in the chromo- somal deletion assay.
Other mutants affecting primosome assembly and
the replication fork were also assayed. Elevated deletion
rates were seen for a temperature-sensitive mutant of
the dnaB helicase and primosome mutants in dnaC and dnaT, assayed at their permissive temperatures. These strains showed a substantial increase in the plasmid de- letion formation as compared to wild-type strains: 20- fold for dnaB107, 11-fold for dnaCl and 15-fold for the double dnaC2 dnaTl2 mutants. The dnaBmutant exhib- ited a large effect in the chromosomal assay with a 130- fold stimulation of deletion rate. Unfortunately, the chromosomal deletion assay could not be used for dnaC
and dnaT mutants due to their extremely poor growth.
Null mutants in the primosome helicase gene PriA
showed a 10-fold increase in the chromosomal deletion
assay and could not be measured in the plasmid assay because of plasmid instability.
We conclude from these studies that aberrant DNA replication via polymerase I11 often results in high rates of deletion between tandem repeated sequences. DNA polymerases I and I1 are not required for deletion for-
mation in E. coli. Recovery of deletions on the chromo-
some is impaired in dnaNand dnaxmutants with defec- tive processivity clamp and clamp loader complex.
Is elevated deletion formation in d m mutants r e d - dependent or -independent? Both recAdependent and -independent pathways contribute to deletion forma-
tion between repeats of the 787-bp size assayed. For
wild-type strains, approximately 70% of the observed deletion is recA-dependent and 30% is recA-independent (LOVETI et al. 1993). The recA-dependent pathway also
uses the recombination genes, recF, recR and rec0 and
may reflect a normal recombinational pathway known
as the RecF pathway (reviewed in SMITH 1989; CLARK
and SANDLER 1994). The genetic basis of the recA-inde- pendent pathway is unknown but has been proposed to involve a replication-slippage (or realignment) mechanism.
We chose three mutants with elevated deletion rates (dnaE486, dnaQ49and dnuBlO7) to determine whether the increased deletion formation was due to recA-depen- dent or -independent processes (Table 3). Plasmid dele- tion rates for dnaE486 mutants were independent of recA, with an 11-fold stimulation in recA- derivatives compared to sevenfold in recA+ derivatives. (The chro- mosomal assay had shown no significant elevation and
so was not tested). Assays of the dna449 mutant strains
showed only a minor effect of recA (twofold elevation
in recA- dnaQ49mutants vs. ninefold elevation in recA+
dnaQ49 mutants for the plasmid assay; 52-fold stimula-
tion in recA- dnaQ49 us. 98-fold in recA+ dnaQ49deriva-
tives for the chromosomal assay). In contrast to the
above, assays of the dnaB107 derivatives showed that most of the large increase seen in the chromosomal
assay was dependent on the recA gene: deletions rates
were 130-fold elevated in recAf but only twofold elevated
TABLE 1
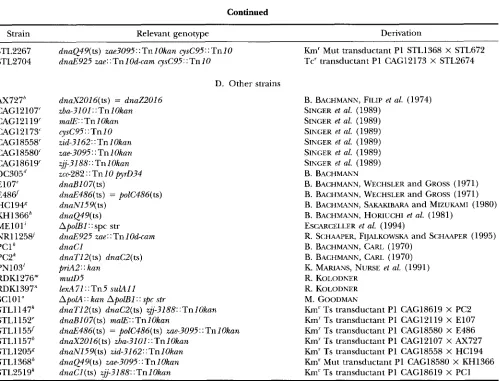
Escherichia coli strains
Strain Relevant genotype Derivation
AB1157" DM49 JC10287
RM4193 STL1270 STL1316 STLl336 STL1818 STL1845 STL1861 STL1866 STL1880 STL1909 STL2107 STLZ 134 STLZ 182 STL2234 STL2314 STL2545 STL'2674 STL2702 STL2706 STL3000
STL695 STL753 STLl324 STL1353 STL1885 STL1887 STL1902 STL1957 STL2077 STL2082 STLZ 109 STL2125 STL2 129 STL2131 STL2 149 STL2181 STL2 186
STL2332 STL2385
STL3022
RM4848 STL672 STL700 STL774 STL1277 STL2128
STL2133
STLZ 174
A. Strains used in plasmid deletion assays
rec+ BACHMANN (1996)
lexA3 A. J. CLARK, MOUNT et al. (1972)
A(srlR-recA)304 CSONKA and CIARK (1979)
holE202: : cat R. MAURER, SIATER et al. (1994)
dnaBl07(ts) malE::TnlOkan Km' Ts transductant P1 STLll52 X AB1157
dnaB107(ts) malE: :TnlOkan A(srlR-recA)304 W Cys' transductant P1 JC10287 X STL1277
ApolBl: : spc str ESCARCELLER et al. (1994)
dnuE486( ts) zae-3095: : Tn 1 Okan Km' Ts transductant P1 STL1155 X AB1157
dnaN159(ts) rid-3162::TnIOkan Km' Ts transductant P1 STL1205 X AB1157
dnaQ49( ts) zae-3095: : Tn 1 Okan K m ' Mut transductant P1 STL1368 X AB1157
priA2: : kan Km' transductant P1 PN103 X AB1157
dnaX2016( t s ) zba-3101: : Tn lOkan Km' Ts transductant P1 STL1157 X AB1157
dnaT12(ts) dnaC2(ts) zji-3188::TnlOkan Km' Ts transductant P1 STL1147 X AB1157
holCl02:: cat Cm' transductant P1 RM4848 X AB1157
sulAI I led71 : : T n 5 Km' transductant P1 RDKl397 X STL2181
dnaE486( ts) zae-3095 : : Tn 1 Okan A(srlR-recA)304 uv' Cys+ transductant P1 JC10287 X STL2174 dnaQ49( ts) zae-3095: : Tn I Okan A(srlR-recA)304 U V Cys+ transductant P1 JC10287 X STL2267
dnaCl (ts) zj-3188: : Tn 1 Okan Km' Ts transductant P1 STL2519 X AB1157
dnaE925 zae: : TnlOd-cam Cm' Km'transductant P1 NR11258 X STL1819
priA2: : kan s u o l l l K m ' transductant P1 STL1866 X STL'2134
dnaE925 zae: : T n l Od-cam A(srlR-recA)304 uv' Cys+ transductant P1 JC10287 X STL2704
mutD5 Pro' Mut transductant P1 RDK1276 X AB1157
S U l A l l Pyr+ Tc' transductant P1 RDK1397 X STL774
B. Strains used in chromosomal deletion assays
l a d : : bla' tetAdup787
A(srlR-recA)304 lacZ: : bla' tetAdup787
dnaBIO7( ts) maE: : Tn 1Okan lacZ: : bla+ tetAdup787 ApolBl : : spc str lacZ: : bla+ tetAdup787
dn&486( ts) zae-3095 : : Tn 1 Okan lacZ: : bla' tetAdup 787 dnaNI59(ts) zid-3162:: TnlOkan lacZ:: bla+ tetAdup787 dnaX2016(ts) zba-3101 ::TnlOkan lacZ:: bla+ tetAdup787 dnaQ49(ts) zae-3095: : Tn 1 Okan lacZ: : bh' tetAdup787 holE202:: cat lacZ:: bla' tetAdup787
priA2:: kan lacZ:: bla' tetAdup787 holClO2: : cat LacZ: : bla+ tetAdup787 lexA3 l a d : : bla' tetAdup787
dnaBlO7(ts) malE: :TnlOkan led3 lacZ:: bla' tetAdup787 dnaQ49(ts) led3 zae-3095::TnlOkan lacZ:: bla+ tetAdup787 ApoL4:: kan lacZ: : bla' tetAdup787
sulAll lacZ: : bla+ tetAdup787
dnaQ49(ts) zae-3095: : TnlOkan A(srlR-recA)304 lacZ: : bla+
s u o l l l lexA71::Tnj lacZ::bla+ tetAdup787
dnaBlO7( ts) malE: : T n l Okan A(srlR-recA)304 lacZ: : bla+ mutD5 lacZ: : bla+ tetAdup787
tetAdup 787
tetAdup787
LOVETT et al. (1993) LOVETT et al. (1993)
Km' Ts transductant P1 STL1152 X STL695 Ap' transductant P1 STL695 X STL1336 Km' Ts transductant P1 STL1155 X STL695 Km' Ts transductant P1 STL1205 X STL695 Km' Ts transductant P1 STL1157 X STL695 Km' Mut transductant P1 STL1368 X STL695 Cm' transductant P1 RM4193 X STL695 Km'transductant P1 PN103 X STL695 Cm' transductant P1 RM4848 X STL695 Ap' transductant P1 STL695 X DM49 Km'Ts transductant P1 STL1152 X STL2125 Km' Mut transductant P1 STL1368 X STL2125 Km' transductant P1 SClOl X STL695 Ap' transductant P1 STL695 X STL2134 uv' Cys+ transductant P1 JC10287 X STL2133
Ap' transductant P1 STL695 X STL2182
U V Cys' transductant P1 JC10287 X STL2128
Pro+ Mut transductant P1 RDK1276 X STL695
C. Intermediate strains derived from AB1157
holCI02:: cat recA430 sr1::TnlO zjg-2086:: kan R. MAURER
cysC95 : : Tn I O Tc' transductant P1 CAG12173 X AB1157
cysC95 : : Tn I O lacZ: : bla+ letAdup 78 7 Tc' transductant P1 CAG12173 X STL695
pyrD34 zcc-282: : Tn I O Tc' Pyr- transductant P1 DC305 X AB1157
dnaB107(ts) malE::TnlOkan cysC95::TnlO Tc' transductant P1 CAG12173 X STL1270
dnaB107(ts) malE::TnIOkan cysC95::TnlO lacZ:: bla+ Km' Ts transductant P1 STLll52 X STL700 dnaQ49( ts) zae-3095 : : Tn IOkan cysC95: : Tn 10 lacZ: : bla' Km' Mut transductant P1 STL1368 X STL700
tetAdup787
tetAdub787
Hyperdeletion in Replication Mutants
TABLE 1
Continued
463
Strain Relevant genotype Derivation
STL2267 STL2704
AX727" CAG12107' CAG12119' CAG12173' CAG18558' CAG18580' CAG18619' DC305" E107' E48d HC 1 94" KH1366" MElO1' NR1125W PC1
PN103' RDKl276" RDK1397" SC101" STLl 147k STLl152' STL1155/ STL1157" STL1205" STL1368" STL251gk PC2k
dna449( ts) zae3095: : Tn 1 Okan cysC95: : Tn I O
dnaE925 zae: ; Tn 1 Od-cam cysC95 : : Tn 10
D. Other strains
dnaX20I 6( ts) = dnaZ2016
zba-3101: : Tn 1 Okan
malE: : Tn 1 Okan
cysC95: : T n 10
zid-3162: : Tn 1 Okan
zae-3095 : : Tn 1 Okan
y'j-3188: : Tn 1 Okan
zcc-282 : Tn 10 pyrD34
dnaB107(ts)
dnaE486( ts) = polC486( ts)
dnaN159(ts) dna449(ts)
ApolBl : : spc str
dnaE925 zae: : Tn I Od-cam
dnaCl
dnaTl2( ts) dnaC2( ts)
priA2: : kan
mum5
lexA 71 ; : T n 5 sul4l I
ApolA: : kan ApolBl : : spc str
dnaT12(ts) dnaC2(ts) zjj"jr188::TnlOkan
dnaBlO7( ts) malE: : Tn 1 Okan
dnaE486( ts) = polC486( ts) zae-3095: : Tn I Okan
dnaX2016( ts) zba-3101: : Tn 1 Okan
dnaN159( ts) zid-3162: : Tn I Okan
dnaQ49( ts) zae-3095: : Tn 1 Okan
dnaCl (ts) zjj-3188: : Tn I Okan
Km'Mut transductant P1 STL1368 X STL672
Tc' transductant P1 CAG12173 X STL2674
B. BACHMANN, FILIP et al. (1974)
SINGER et al. (1989)
SINGER et al. (1989)
SINGER et al. (1989)
SINGER et al. (1989)
SINGER et al. (1989)
SINGER et al. (1989)
B. BACHMANN
B. BACHMANN, WECHSLER and GROSS (1971)
B. BACHMANN, WECHSLER and GROSS (1971)
B. BACHMANN, SAKAKIBARA and MIZUKAMI (1980)
B. BACHMANN, HORIUCHI et al. (1981)
ESCARCELLER et al. (1994)
R. SCXAAPER, FIJALKOWSKA and SCHAAPER (1995)
B. BACHMANN, CARL (1970)
B. BACHMANN, CARL (1970)
K. MARIANS, NURSE et al. (1991)
R. KOLODNER R. KOLODNER M. GOODMAN
Km'Ts transductant P1 CAG18619 X PC2
Km'Ts transductant P1 CAG12119 X E107
Km'Ts transductant P1 CAG18580 X E486
Km'Ts transductant P1 CAG12107 X AX727
Km'Ts transductant P1 CAG18558 X HC194
Km'Mut transductant P1 CAG18580 X KH1366
Km'Ts transductant P1 CAG18619 X PC1
" Genotype of AB1 157, STL695 and their derived strains, unless otherwise indicated, includes F- X- hisG4 argE2 leuB6 A(gpt-
I' Genotype of AX727 and its derivative includes F- thi lac rpsL rpsL.
'All CAG strains are derived from MG1655 and their genotype includes F- X-.
"Genotype of DC305 includes F- galK2 mal41 xyl-7 mtl-2 rpsL118.
'Genotype of E107 and derivative includes F- thr-1 leuBGJhwl21 supE44 X- @Dl thyA6 rspL67 thildeoC1.
/Genotype of E486 and its derivative includes F- thr-1 leuB6 tonA2l lacy1 supE44 X- @Dl thyA6 rpsL67 thil deoC1 met89.
"Genotype of HC194 and its derivative includes Hfr tow422 ompF627 reoll thyA148 metBl T T .
'Genotype of ME102 includes F- X- tonA2l thrl leuB6 lacy1 supE44 @Dl thil.
I Genotype of NR11258 includes F- aru thi A(pro-lac) mutL:: Tn5.
'
Genotype of PN103 includes HfrC X-.proA)62 thr-1 thi-1 rpsL31 galK2 lacy1 ara-14 xyl-5 mtl-1 kdgK5l supE44 tsx-33 @Dl mtl-1 rac-.
Genotype of KH1366 and its derivative includes Hfr metD88 Alac-6 tsx-7 X- srl-8 reUl spoT1 metB1.
Genotype of PC1, PC2 and their derivatives includes F- leuB6 X- thyA47 qsL153 deoC3.
Genotype of RDK1276 includes lacZ trpA metE.
Genotype of RDKl397 includes F- leuB6 thr-1 p o met x y l mtl gal ara.
"Genotype of SClOl includes F+ A(ga1-bio) thi-1 rel4l spoT1.
rates were also somewhat recA-dependent, with 20-fold elevation in recA' dnaB107strains and 4.5-fold elevation
i n recA- dnaB107 strains. We conclude that both recA-
d e p e n d e n t and -inde pe ndent d el e ti o n form a t i on can
be induced by aberrant DNA replication and that indi-
vidual replication mutants differ in the mechanism by
which deletion formation is stimulated.
Effect of the SOS response on deletion forma- tion: The SOS response in E. coli is the coordinated
induction of a set of genes whose function is to p r o m o t e
survival after production of replication-blocking lesions
i n DNA. Some replication mutant strains such as dnuQ
and PriA exhibit constitutive expression of the SOS re-
sponse (NURSE et al. 1991; SLATER et al. 1994) and it
is u n k n o w n w h a t t h e c o n s e q u e n c e o f t h i s m a y be o n observed tandem deletion rates.
Induction of the SOS response was neither necessary
nor sufficient to induce deletion formation (Table 4).
464 C. J. Saveson and S. T. Lovett
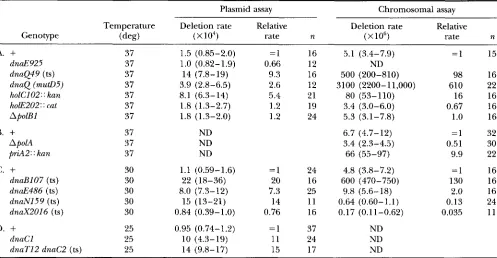
TABLE 2
Deletion rates in red' background
Plasmid assay Chromosomal assay
Temperature Deletion rate Relative Deletion rate Relative
Genotype (deg) ( ~ 1 0 ~ ) rate n ( X
lo6)
rate nA.
+
37 1.5 (0.85-2.0) = I 16 5.1 (3.4-7.9) = I 15dnaE925 37 1.0 (0.82-1.9) 0.66 12 ND
dnaQ49 (ts) 37 14 (7.8-19) 9.3 16 500 (200-810) 98 16
dnaQ (mutD5) 37 3.9 (2.8-6.5) 2.6 12 3100 (2200-11,000) 610 22
holCl02 : : kan 37 8.1 (6.3-14) 5.4 21 80 (53- 110) 16 16
holE202: : cat 37 1.8 (1.3-2.7) 1.2 19 3.4 (3.0-6.0) 0.67 16
ApolBl 37 1.8 (1.3-2.0) 1.2 24 5.3 (3.1-7.8) 1
.o
16B.
+
37 ND 6.7 (4.7-12) =1 32ApolA 37 ND 3.4 (2.3-4.5) 0.51 30
priA2: : kan 37 ND 66 (55-97) 9.9 22
c.
+
30 1.1 (0.59-1.6) =1 24 4.8 (3.8-7.2) =1 16dnaBl07 (ts) 30 22 (18-36) 20 16 600 (470-750) 130 16
dnaE486 (ts) 30 8.0 (7.3-12) 7.3 25 9.8 (5.6-18) 2.0 16
dnaN159 (ts) 30 15 (13-21) 14 11 0.64 (0.60-1.1) 0.13 24
dnaX2016 (ts) 30 0.84 (0.39-1.0) 0.76 16 0.17 (0.11-0.62) 0.035 11
D.
+
25 0.95 (0.74-1.2) = I 37 NDdnaCl 25 10 (4.3-19) 11 24 ND
dnaTl2 dnaC2 (ts) 25 14 (9.8-17) 15 17 ND
All strains are derivatives of AB1157 and are described in Table 1. The strains in A and B were assayed at 37" and the strains in C and D were assayed at permissive temperatures of 30 and 25", respectively. The strains in A, C and D were grown on LB medium, while the strains in B were grown on minimal complete medium. All mutant strains were assayed in parallel with the wild-type control strains AB1157 or STL695. Rates were calculated by the method of the median (LEA and COULSON 1949) and represent an estimation of the number of deletion events that occurred in the population, calculated from the observed median number of Tc' colonies for the number of independent cultures ( n ) , as described in the MATERIALS AND METHODS. Numbers in parentheses correspond to 95% confidence values for the measured rates. Relative rates were determined by comparison to the control strain assayed under the same growth conditions. ND, not determined.
causes constitutive expression of SOSregulated genes, However, the SOS response may augment the deletion
showed wild-type levels of deletion formation in both rate in this strain somewhat, which accounts for the
the plasmid or chromosomal assays. Furthermore, the twofold decrease in rates seen in both recA and ZexA
large increase in deletion formation promoted by E mu- Ind- dnuQmutant derivatives relative to rec+ kx+ dnuQ
tations does not require the SOS response with the strains. In contrast to dnaQ a substantial proportion of
double mutant dnaQ49 kxA3 (Ind-) showing a 47-fold increased deletion formation in
dnuBlO7
strains wasincrease of deletion formation in chromosomal assays. ZexA-dependent. Deletion rates on the chromosome
TABLE 3
Deletion rates in red derivatives
~~~ ~
Plasmid assay Chromosomal assay
Temperature Deletion rate Relative Deletion rate Relative
GenotvDe (deg) ( ~ 1 0 ~ ) rate n (X106) rate n
A. A recA304 37 0.69 (0.38-1.3) =1 16 5.2 (2.7-5.7) =1 16
ArecA304 dnaQ49 (ts) 37 1.2 (0.48-2.3) 1.7 19 270 (60-610) 52 12
B. ArecA304 30 0.51 (0.28-0.56) =1 23 5.3 (2.9-7.6) =1 16
ArecA304 dnaB107 (ts) 30 2.3 (1.1-5.7) 4.5 19 9.5 (6.3-1.7) 1.8 16
ArecA304 dnaE486 (ts) 30 5.4 (2.3-7.8) 11 23 ND
All strains are derivatives of AB1157 and are described in Table 1. The strains in A were assayed at 37" and the strains in B were assayed at a permissive temperature of 30°, in parallel with ArecA304 control strains JC10287 or STL753. Rates were calculated for the number of independent cultures ( n ) as described in MATERIALS AND METHODS. Values in parentheses correspond
465 Hyperdeletion in Replication Mutants
TABLE 4
Deletion rates for kxA derivatives
Plasmid assay Chromosomal assay
Temperature Deletion rate Relative Deletion rate Relative
Genotype (deg) ( ~ 1 0 ' ) rate n ( X 106) rate n
Zed3 (Ind-) 37 ND 4.3 (2.2-8.6) 0.84 16
Zed3 (Ind-) dnaQ49 37 ND 240 (91
-
280) 47 12Zed3 (Ind-) dnuBl07 (ts) 30 ND 140 (47-350) 29 15
sulAll Zed71 (De0 37 1.7 (1.1-2.8) 1.1 20 12 (8.3-22) 2.4 16
All strains are derivatives of AB1157 and are described in Table 1. The strains were assayed in parallel with the wild-type
control strains AI31157 or STL695. Rates were calculated for the number of independent cultures (n) as described in MATERIAIS
AND METHODS. Values in parentheses correspond to the 95% confidence limits for the rates. Relative rates were determined by
comparison to the control strain assayed under the same growth conditions. ND, not determined.
S U l A l l 37 1.1 (0.76-1.6) 0.73 20 9.2 (5.7-14) 1.8 12
were 29-fold elevated over wild-type strains for Zed3
dnaBlO7 compared to 130-fold for lex+ dnaBlO7 deriva- tives. Mutations in the DnaB fork helicase and primo- some protein therefore promote deletions, in part, via
a mechanism requiring induction of the SOS response.
Analysis of plasmid deletion products reflects two
types of Red-independent deletion formation: There are several mechanisms that may give rise to deletion events between tandem duplications. Unequal sister- chromosome exchange can lead to deletion formation; such events fuse circular replicons and can be detected in the plasmid assay by the formation of dimeric plas- mid deletion products. Other mechanisms of deletion formation, such as slipped mispairing, are expected to produce only monomeric products (see Figure 4).
Examination of the plasmid deletion products for their monomeric or dimeric structure can therefore be informative about the particular deletion mechanism
in play. However, product analysis in recA+ strains is
confounded because of the occurrence of recombina- tion between plasmids before and after the deletion event. For this reason, we have examined products in the recA strain background, where there is no detectable plasmid recombination (and plasmid dimerization) in
the absence of the deletion event. Among deletion
products in recA strains, 40% were dimeric plasmid
products that represent intramolecular recA-indepen-
dent sister-chromosome exchange. The remaining 60%
of products were monomeric, formed presumably by
slipped mispairing ( LOVETT et al. 1993).
To determine whether mutations in the replication machinery affect one of these mechanisms specifically, we analyzed the plasmid deletion products of selected
replication mutants (Table 5). Among dnaE486 ArecA
mutants assayed at their permissive temperature, where an 11-fold stimulation of recA-independent deletion is observed, proportionately more products are found in
the dimeric form. Conversely, in dnaQ49 ArecA strains,
where recA-independent deletion events are stimulated
twofold relative to dnaQ+ ArecA strains, more products
are found in the monomeric form. The transformation
efficiency of monomer and dimer forms into these mu-
tants, with and without a coresident monomer plasmid (data not shown), confirmed that the skewed product
distribution is not merely due to a change in the estab-
lishment or maintenance of various plasmid forms.
Rather, these results suggest that polymerase mutation
dnaE486 increases the likelihood of a dimer-producing deletion event, such as sister-chromosome exchange
(see Figure 4). The editing subunit mutation dnaQ49,
on the other hand, increases the likelihood of mono- mer-producing mechanism such as slipped mispairing on the same strand (as shown in Figure 4). The antimu-
tator polymerase mutation dnaE925 and helicase muta-
tion dnaBl07have no effect on the distribution of dele-
tion products. Because dnaB107 caused a 4.5-fold
stimulation of recA-independent deletion, both dimer- and monomer-producing pathways must be equally en- hanced.
DISCUSSION
We have demonstrated that mutations in many repli- cation genes affect deletion events at tandem repeats. For most of the mutant strains examined, we observed increased deletion of repeats both carried on a
multicopy plasmid and on the E. colichromosome. How-
ever, for dnaQ and dnaB mutants there were much
larger increases in chromosomal as compared to plas- mid deletion rates. This discrepancy may be due to
several reasons. (1) The differences between plasmid
and chromosomal data may reflect a real difference in
the way that replication occurs on the plasmid us. the
chromosome. Both the chromosome and the plasmid
depend on Pol I11 for normal replication although the
structure of the replication fork could differ in some way [there are differences in the genetic requirements
for initiation and primosome assembly (KORNBERG and
BAKER 1992)l. (2) The chromosome may be subject to
466 C. J. Saveson and S. T. Lovett
TABLE 5
Deletion product analysis for dnuB, dnaE and dnaQ mutants
Growth
temperature Dimer
Genotype products
A. A recA304 37 32/82 (39)
ArecA304 dnaQ49 (ts) 37 15/93 (16)
ArecA304 dnaE925 37 32/82 (39)
B. ArecA304 30 48/140 (34)
ArecA304 dnaBlO7 (ts) 30 39/107 (36)
ArecA304 dnaE486 (ts) 30 54/87 (62)
All strains are derivatives of AB1157 and are described in
Table 1. The strains in A were grown at 37" and in B at
30". Dimer product ratios represent the number of deletion products that were dimeric plasmids (sister-chromosome ex-
change events) out of the total number of plasmid deletion
products examined for each strain, with the percentage in
parentheses. The alternative product was monomeric plas-
mid, equivalent to pBR322.
x'
analyses showed that the distri-butions of products in strains dnaE486 and dnaQ49 were sig-
nificantly different from control strain ArecA304.
on plasmids. For example, the plasmid lacks
x
se-quences that could stimulate recombinational reactions on the chromosome. (3) Differences observed may re- flect relative recovery of products. For multicopy plas- mids, deletion intermediates may be selectively lost. In- deed, plasmid loss may be magnified in mutants that impair replication, causing reduced recovery of plasmid
deletion products. Determination of plasmid copy num-
ber (data not shown) suggested that dnaB and dnaC
dnaT mutants, in particular, have two- to fivefold low-
ered levels of plasmid DNA, which could lead to an
underestimate of plasmid deletion rates in these strains.
The other replication mutants tested here were not
significantly affected in this analysis (data not shown).
In two cases, the dnaN and dnaX mutant strains, we
observed seemingly contradictory results: no effect or an increase in deletion events for repeats on the plas- mid but a large decrease in deletions of the same re- peats on the chromosome. These mutations affecting the processivity clamp may truly affect tandem repeat
deletion in opposite manners on a plasmid us. a chro-
mosome replicon. An alternative explanation, which we prefer, is that, in these mutant strains, chromosomal deletion products may be less likely to be recovered. For instance, aberrant or incomplete replication may
initiate deletion formation on both the plasmid and
the chromosome. However, on the chromosome, the deletion intermediate may fail to be processed into a viable deletion product. Because the plasmid presents a shorter replication template, it is possible for ineffi- cient Pol I11 replication or uncoupled leading and lag- ging strand replication to be completed or replaced by repair replication via Pol I or 11. On the much larger chromosome, the same situation may lead to failure to
complete replication and loss of the potential deletion product.
Of E. coli's three known DNA polymerases, only func-
tions associated with Polymerase 111, responsible for
chromosomal replication, affected deletion at tandem repeats in our experiments. This includes mutations in
the a polymerase (dnaE gene) and E exonuclease
(dnaQ) subunits of the core enzyme as well as mutations
in subunits of the y complex (dnaX, hoZC) and the ,B
clamp (dnaN). Mutants in priA, dnaB, dnaC and dnaT
with defects in the primosome or primosome assembly also exhibited high deletion rates. Another mutant of
Polymerase 111, the antimutator dnaE (FIJALKOWSKA and
SCHAAPER 1995) did not show altered deletion rates. Deletions of genes encoding Polymerases I and I1 had no effect on deletion rates in otherwise wild-type strains. This result does not, however, exclude the possibility that these polymerases also contribute in a minor way
to deletion formation. The ssb-113 mutation of single-
strand DNA binding protein had been previously shown
to elevate deletion formation fourfold in the same plas-
mid assay as used here (LOVETT et al. 1993). This mutant
protein has normal DNA binding properties and is be-
lieved to be defective in protein-protein interactions
(CHASE et al. 1984) with components of the replication complex.
We believe that deletion formation is stimulated in these mutants by aberrant DNA replication rather than other indirect effects. The phenotype of elevated dele- tion frequency is not correlated with slow growth. Dele- tion rates in wild-type were found to be largely indepen- dent of growth medium and temperature. Whereas some slow-growing replication mutants showed elevated deletion rates (dnaB), others showed essentially wild-
type deletion rates despite poor growth (poZA)
.
In addi-tion, our results suggest that induction of the SOS re-
sponse does not itself stimulate deletion formation nor
is it required for the elevated rate seen in some hyperde-
letion mutants, as in the case of the dnaQ mutant. Al-
though this suggests that aberrant replication in a vari- ety of replication mutant strains can lead to enhanced deletion formation at tandem repeated sequences, this does not necessarily mean that deletion events in wild- type strains occur strictly during the process of replica- tion. Recently, we have presented genetic evidence that most deletions formed between 101-bp tandem repeats occur during, or shortly after, DNA replication because they can be subject to correction dependent on hemi-
methylation (LOVETT and FESCHENKO 1996). Moreover,
the reduced recovery of chromosomal deletion prod- ucts in dnaNand dnaxmutants also implies that replica- tion is an essential component of the deletion process.
More detailed analysis of several selected mutants
(dnaB, dnuE and dna4) suggested that aberrant replica- tion can stimulate deletion through different pathways. The hyperdeletion phenotypes for polymerase mutants
Hyperdeletion in Replication Mutants
whereas that for dnaR helicase mutant was primarily rPcAdependent. Also, a functional SOS response was necessary to see the full stimulatory effect of dnaR muta- tions on deletion formation, in contrast to dnuQ. The mutant polymerase must therefore stimulate a recA and Zed-independent deletion pathway whereas the mutant DnaB helicase induces primarily a recA and Zeddepen- dent recombinational deletion pathway.
Analysis of the structure of plasmid deletion products has revealed that there are two mechanisms of RecA- independent deletion formation. Deletion formation on circular replicons results in both monomeric and dimeric forms. According to replicational models of deletion, these products reflect distinct mechanisms: simple slippairing, resulting in monomer replicon
products, and sisterchromosome slippairing, resulting in dimeric replicon products (Figure 4). The distribu- tion of products in the dnaQ49 strain was skewed to monomers and that in dnaE486 strains was skewed to dimers, relative to the proportion observed for wild- type strains. Product analysis in a dnaB strain revealed a distribution similar to wild type. We conclude that aberrant replication can stimulate deletions in both a RecAdependent and -independent manner, and via both simple slippage and the sisterchromosome ex- change mechanisms. Different replication mutants dif- fer in the mechanism by which these deletions occur.
The correlation of hyperdeletion phenotypes with specific biochemical defects in replication fork propa- gation may shed light on the mechanisms of deletion formation. However, more exhaustive genetic and bio- chemical analysis will be necessary before clear conclu- sions can be drawn. For many of the mutants examined here, the specific consequences of the mutation on the structure or function of the replication complex in vivo is not well understood. The generality of phenotypic effects is also not clear because only one mutant allele was examined in most cases. Moreover, many of our assays were performed at “permissive” temperatures, where the function of temperature-sensitive compo- nents of the replication machinery may be impaired in subtle ways. It is not known whether observed effects on deletion formation are genetically recessive or domi- nant, and therefore due to a missing or an altered activ- ity. Deletion events occur rarely, in a small fraction of cells, and therefore may not necessarily correlate to the biochemical defects determined for the cell population
as a whole. Nevertheless, in theory, the biochemical functions affected by these hyperdeletion replication mutants allow us to speculate on several ways by which deletion formation may be stimulated.
Some mutants may encourage deletion by promoting the persistence of single-strand DNA in the replication fork. Mutations that adversely affect replication on only one strand may cause extended stretches of single- stranded DNA to accumulate during replication. Muta- tions affecting the primosome complex (e.g., dnaB,
467
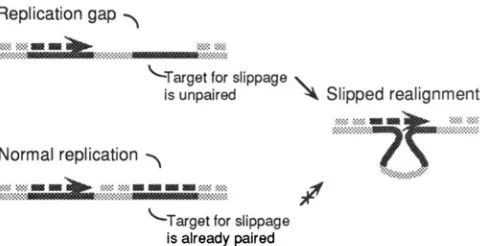
Replication gap
“:::z&Rw>>>>>.
A,. ,. :.:*, %... ..x.< ,..% <Target for slippageis unpaired
\
Slipped realignment.:,:.s.:.z<. Ey >>y X?> :*
. . . c<:sz.*:::::
Normal replication
y
.
:
:
#
,
I
,
.
.
.
.
.
.
I
I
I
.
.
.
.
.
.
<Target for slippage is already paired
FIGURE 5.-Persistence of singlestrand DNA promotes slipped realignment. If a replication gap encompasses a copy of the tandem repeat, the nascent strand is free to realign at the second repeat. This slipped realignment, if unrepaired, will give rise to deletion. During normal replication, no such singlestrand DNA is available for alternative pairing and slipped alignments are discouraged.
dnaC, dnaTand p i A ) could be expected to disturb rep- lication specifically on the lagging strand, either by un- coupling leading and lagging strand replication or by leaving gaps of unreplicated ssDNA on the lagging strand.
Persistence of single-strand gaps could promote a RecAdependent mechanism of deletion formation. Daughter-strand gaps caused by replication-blocking le- sions in DNA, such as UV-induced pyrimidine dimers, are filled by recombinational postreplication repair (Figure 2 ) . This repair process requires RecA protein in order to recombine nascent strands across the repli- cation fork (RUPP et dl. 1971). Alternatively, a stalled replication fork may be broken and then reestablished by RecAdependent recombination with its sister chro- mosome ( h A I et al. 1994; KUZMINOV 1995a,b) (Figure 3). One or both of these mechanisms may be induced by the dnaR mutation which stimulated deletion forma- tion in a recA- and lexAdependent fashion. The dnaR mutation may block unwinding of the fork or leave daughter-strand gaps by disrupting primosome func- tion, thereby inducing the recombinational repair pro- cesses that culminate in deletion.
RecA-independent deletion formation may also be induced by the accumulation of single-strand DNA in the replication fork (Figure 5). Strand realignment would be disfavored if the secondary pairing site had already been replicated to duplex DNA; available tar- gets of single-stranded DNA template would promote slippage and mispairing either on the same strand or across sister strands. A minor component of the stimula- tion by dnuR on deletion formation was recA-indepen- dent and may occur by this mechanism.
468 C. J. Saveson and S. T. Lovett
Displacement
of
nascent strand during replication4
.:.:.:.:.:.:.:.~.x.:.:.:.:.: k??: ::::::: ::;::::
=
*e
:C*S.X.h >x+x.:.:.:.x.:.:.:.:.>J
Slipped realignment
1
1
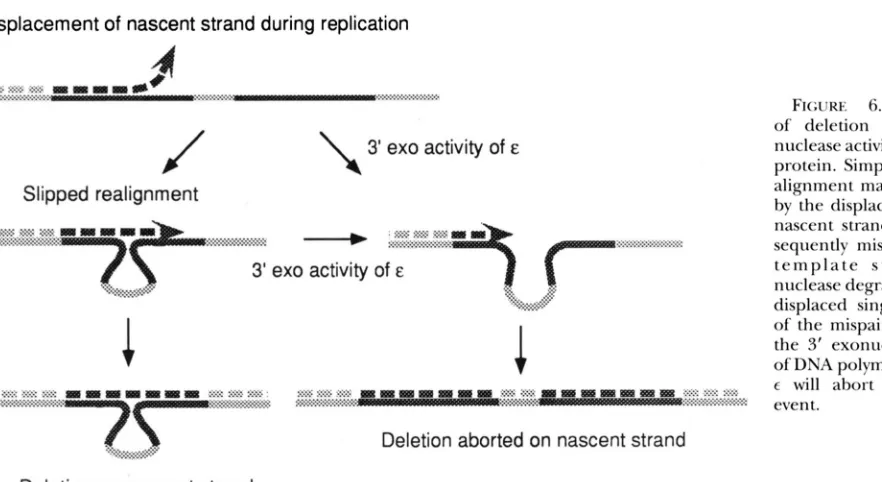
FIGURE 6"Prevention of deletion by the exo- nuclease activity of e ( d m @
protein. Simple slipped re- alignment may be initiated by the displacement of the nascent strand, which s u b
sequently mispairs with its t e m p l a t e s t r a n d . Exo- nuclease degradation of the displaced single strand o r
of the mispaired strand by the 3' exonuclease activity of DNA polymerase subunit
E will abort the deletion
event.
Deletion on nascent strand
ciation of the polymerase could free the nascent DNA strands and allow their translocation across the fork or on their template strands to produce slipped mispair- ing. Mutations in the a subunit of the polymerase may stimulate Red-independent recombination via this mechanism.
Other mutants in the
p
clamp and clamploading/ recycling machinery y complex may stimulate deletion formation by decreasing processivity of the polymerase. We would expect mutations in the clamp or mutations to the polymerase that hinder its interactions with the clamp, to inhibit the processivity of the enzyme com- plex, thereby increasing the incidence of deletions. As with the primosome, one might expect defects in the clamp loader to affect lagging-strand synthesis more than leading-strand synthesis, slowing replication or leaving single-stranded gaps. However, in some cases, deletions on the chromosome may not be recoverable because of failure to reinitiate and complete replica- tion.Replication mutations may also stimulate deletion by allowing slipped mispairing intermediates to persist. We observed that both the dnaQ49 and mutD5 mutation increased deletion events. At least in the case of dnuQ49, this increase was via a recA-independent mechanism. This may be via simple slipped mispairing, as we ob- served proportionately more monomer deletion prod- ucts in dnaQ49mutants. Figure 6 illustrates that, during normal replication, the nascent strand may be displaced from its template. The E subunit of Pol
I11
may degradethis displaced single strand from its 3' end and thereby prevent slipped pairing events from occurring. Alterna- tively, E may degrade the 3' end of the nascent strain
in the slipped alignment with its template and abort the deletion process. Although these scenarios postu-
late that it is the loss of the exonuclease activity that is responsible for the dnaQeffects, E has also been shown
to modulate a and the processivity of the polymerase (STUDWELL and O'DONNELI. 1990) and this property may influence the deletion process, as described above. Our observations that mutations in the replication machinery may greatly stimulate genetic rearrange- ments may have implications for human genetic dis- ease. Rearrangements between tandem repeated se- quences or genes are a source of genetic mutation in humans (MEUTH 1989; K R A W C ~ A K and COOPER 1991;
HU and WORTON
1992; NEISON 1993). It is likely that aberrant replication in humans, as in other organisms, can also promote deletion events. Mutations in the anal- ogous replication functions of human cells to those we have investigated here may well predispose certain indi- viduals for more frequent genetic rearrangements and genetic disease.We thank PAOIA DRAPKIN and ~ . E R GI.UCKMAN for preliminary
work o n this project and VIADIMIR FESFIENKO for construction o f the
poL4 mutant strain. We are indebted to the following individuals for
providing strains: B. BACHMANN of the P;. coli Genetic Stock Center,
A. J. CIARK, M. GOODMAN, C. GROSS, R. KOI.ODNF,R, K. MARIANS, R.
MAURER and R. SCIIAAPER. We thank the anonymous reviewer for
suggesting the statistical method for setting 95% confidence limits for
rate determinations. This work was supported by U.S. Public Health
Service grants R 0 1 GM-51753 to S.T.L. and T32 GM-07122 to C:J.S.
LITERATURE CITED
AIBERTINI, A. M., M. HOFER, M. P. C h o s and J. H. MII.I.ER, 1982 O n the formation o f spontaneous deletion: the importance of short sequence homologies in the generation of large deletions.
Cell 2 9 319-328.
ARAI, K., and A. KORNRERG, 1981 Unique primed start of phage
4x174 DNA replication and mobility of the primosome in a
direction opposite chain synthesis. Proc. Natl. Acad. Sci. USA