Derivation and Variability. (Under the direction of David Threadgill & James Mahaffey). Induced pluripotent stem cells (iPSCs) are differentiated cells that have been
reprogrammed back into an undifferentiated embryonic stem cell like state with the ability to differentiate into any cell type of the adult organism. Induced pluripotent stem cells offer a potentially unlimited source of patient specific embryonic stem cell like (ESC-like) cells that could be used for both research and therapeutics. Although they are being used extensively, there are limited studies on the effect of genetic background on the ability to derive induced pluripotent stem cells. Embryonic stem cell research suggests that genetic background has a profound effect on the ability to derive embryonic stem cells, with some genetic backgrounds resistant to ESC derivation under standard culture conditions.
Standard culture conditions are defined as serum-based media supplemented with LIF protein and was initially used for the derivation of ESCs from various mouse 129 sub-strains. The inability to derive ESCs under standard conditions is termed permissive. For non-permissive mouse strains, the epiblast of the post implantation egg cylinder stage, which gives rise to epiblast stem cells (EpiSCs), is the earliest developmental time point that stem cells can be readily derived. Epiblast stem cells are notably more developmentally primed and are thus less ideal for scientific research and therapeutics than embryonic stem cells.
We tested the ability of eight genetically diverse inbred mouse strains to derive iPSCs and found that six strains are permissive to iPSC derivation, whereas two strains, the
reprogramming for a minimum of seven days to derive ESC-like iPSCs from non-permissive strains.
Previous studies report that ESCs derived from non-permissive strains using GSK3B and MEK inhibitors are metastable. In those studies the removal of the inhibitors resulted in the transition of the cells to an EpiSC fate, dependent on epiblast stem cell culture conditions. Here, iPSCs derived using the 2iS technique from non-permissive strains can be stably removed from GSK3B and MEK culture conditions. They do not transition to an EpiSC fate. Rather, they transition to a transcriptionally heterogeneous state resembling that of ESCs derived in standard culture conditions.
by
Tiffany Aimeé Garbutt
A dissertation submitted to the Graduate Faculty of North Carolina State University
in partial fulfillment of the requirements for the degree of
Doctor of Philosophy
Genetics
Raleigh, North Carolina 2016
APPROVED BY:
_______________________________ _______________________________ David Threadgill James Mahaffey
Committee Co-Chair Committee Co-Chair
_______________________________ _______________________________ David Aylor Dahlia Nielsen
DEDICATION
To our son, Benjamyn Neil Garbutt
BIOGRAPHY
Tiffany Aimeé Garbutt (née Bernier) was born on October 2, 1988 on St. Thomas, United States Virgin Islands (USVI), to James and Theresa Bernier. She is a third-generation Virgin Islander and is proud of her diverse Caribbean roots. Her parents are hard workers, who instilled in her the values of dedication, determination, and responsibility. They also kindled a light to dream big and to use those values to achieve your dreams.
She graduated valedictorian from Sts. Peter and Paul Catholic School and attended the University of the Virgin Islands (UVI). She began her undergraduate degree intending to pursue humanities. However, she took a biology class wherein she learned about genetics and became instantly intrigued. She had family members who had been affected by deafness and hemophilia and her childhood best friend had cystic fibrosis. She felt empowered by the ability to conceptualize these known genetic diseases and switched her major to biology.
She worked throughout her college career in various positions. However, her most impactful experience was participating in the Emerging Caribbean Scientist Program, where she conducted weekly hands on research in green sea turtles and plants. She also participated in the Interdisciplinary Summer Undergraduate Research Program at the University of Iowa, where she discovered and researched a large deletion in a pediatric patient’s genome.
ACKNOWLEDGMENTS Academic Thank You!
Thank you to my committee; I have learned valuable lessons from each of you. A special thank you to my advisor David Threadgill for his guidance and support throughout these years. I appreciate that you have always been there for me, even from afar as a listening ear and guide. A special thank you to David Aylor, who graciously adopted me into his lab and served as a surrogate mentor. I appreciate your guidance and that you always made time for me. A special thank you to James Mahaffey for reminding me to see the new scientific story with each finding and to be excited about the twists and turns of scientific data.
I am truly grateful to the Threadgill and Aylor labs. I am lucky to have been a member of both of these great groups and to have made awesome friendships.
Thank you to Kranti Konganti and Thomas Konneker for being patient guides in my foray into bioinformatics. Thank you to Andrew Hillhouse for his help with sequencing and single cell capture. Thank you to Laura Reinholdt, Jacob Hanna, Ludovic Vallier, and Alice Jouneau for generously gifting some of the cells used in this study.
Thank you to my undergraduate students, Alexis Jones and Drake Phelps, for all their hard work and assistance throughout this project. It was my pleasure to work with you both. I know you will continue on to do great things.
Thank you to the Program in Genetics staff, both past and present for all their help and for knowing just what to do. Lastly, thank you to the NSF Graduate Research
Personal Thank You!
Thank you to God. I am not sure I would have made it this far without faith. It has given me belief even in my weakest moments that answers can and will be found.
Thank you to my husband, Bayley K. Garbutt. You understand this journey more than anyone else. Thank you for going on this adventure with me, for listening to me, for
believing in me, and for motivating me. Your encouragement and support has kept me going even when I felt like giving up. You are my partner and my best friend. I love you and can’t wait to be there at your dissertation!
Thank you to my parents, James and Theresa Bernier for instilling in me the values of hard work, dedication, determination, resiliency, and responsibility. These qualities have been the cornerstone to finishing this degree. Thank you for believing in me and supporting my dreams. More practically, thank you for feeding my hungry little family when we are too busy to cook and reminding me to take breaks.
Thank you to my mother in law, Yvette R. Garbutt I truly appreciate your help, support, love, and encouragement.
Thank you to my siblings, James Bernier Jr and Tina King, time spent with you and your families has brought me much relief and joy. Thank you to the rest of my family.
TABLE OF CONTENTS
LIST OF TABLES ... viii
LIST OF FIGURES ... ix
INTRODUCTION ...1
STEM CELLS ...1
CELLULAR REPROGRAMMING ...2
Early Cellular Reprogramming ...2
Single Cell Nuclear Transfer ...3
Cell Fusion ...5
Transcription Factor Reprogramming ...7
INDUCED PLURIPOTENT STEM CELLS ...8
Discovery ...8
Advantages / Benefits ...9
iPSC & ESC Equivalency ...12
Methods of Reprogramming ...15
Functions of the Four Factors ...17
Models of Reprogramming ...19
EPIGENETIC REPROGRAMMING ...22
Epigenetic Model ...22
OKSM Factors ...23
Histone Modifying Enzymes ...25
Chromatin Architecture ...27
DNA Methylation ...28
MOUSE AS A MODEL ...30
Brief History & Practicality of Mouse as a Model ...30
Mouse Embryonic Development ...34
Non-permissive vs. Permissive ...37
ESC vs. EpiSCs in vitro ...41
Embryonic Stem Cell Heterogeneity ...44
CONTRIBUTION OF THIS STUDY ...48
REFERENCES ...50
GENETIC BACKGROUND INFLUENCES iPSC DERIVATION ...71
ABSTRACT ...71
INTRODUCTION ...72
RESULTS ...75
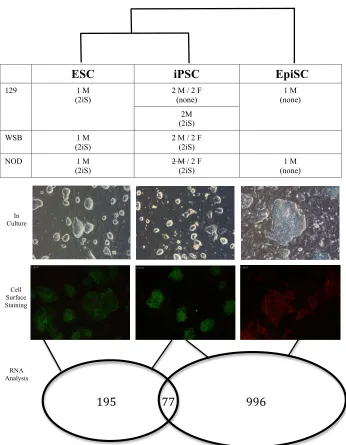
NOD/LtJ and WSB/EiJ are non-permissive to iPSC derivation in standard conditions ..75
NOD non-permissiveness is recessive ...82
2iS media derives ESC-like iPSCs from fibroblasts of non-permissive mouse strains ...86
NOD male iPSCs are transcriptionally distinct from other iPSCs ...96
DISCUSSION ...113
METHODS ...120
REFERENCES ...134
DERIVATION OF STABLE EMBRYONIC STEM CELL-LIKE, BUT TRANSCRIPTIONALLY HETEROGENEOUS IPSCS FROM NON-PERMISSIVE MOUSE STRAINS ...147
ABSTRACT ...147
INTRODUCTION ...149
RESULTS ...151
Short 2iS treatment reprograms cells to an early heterogeneous ESC-like state ...151
2iS maintains a metastable homogenous state ...163
There is a difference between cells from homogneous populations in 2iS and cells from heterogenous populations not in 2iS ...166
CD40+ cells were not captured in the random selection of single cells ...182
PECAM1+ cells and dual expressing cells are more like ESCs than EpiSCs ...188
FOXO1 and MYC are significantly different between PECAM1+ and CD40+ cells ...194
DISCUSSION ...197
METHODS ...202
REFERENCES ...214
CONCLUSION ...224
PRELIMINARY FINDINGS ...224
RATIONALE TO OVERCOMING NON-PERMISSIVENESS ...224
RESULTS OF 2IS TREATMENT ...226
STABILITY OF 2IS TREATMENT ...229
SINGLE CELL HETEROGENEITY ...232
HETEROGENEITY & HOMOGENEITY IN THE DEVLEOPMING EMBYRO ...236
CELL POTENTIAL MODEL ...239
FINAL THOUGHTS ...242
REFERENCES ...244
APPENDICES ...250
APPENDIX A CHAPTER 2 SUPPLEMENTAL DATA ...251
LIST OF TABLES
Chapter 2
Table 1 Antibodies tested to determine the best markers of ESC and EpiSC states ....133
Chapter 3 Table 1 The four 2iS treatment groups for the NOD background ...167
Table 2 Conversion from percentage to the number of live and dead cells ...185
Table 3 Converted percentages for the repeat of the flow sorting experiment ...186
Table 4 Significant functional groups associated with PECAM1+ cells ...191
LIST OF FIGURES
Chapter 1
Figure 1 Pluripotency and mouse development ...36
Figure 2 Difference between permissive and non-permissive strains ...39
Figure 3 The mouse embryonic stem cell state is a derived state ...40
Figure 4 The LIF pathway activates pluripotency and differentiation pathways ...43
Figure 5 FGF2 and ACTIVIN maintain mouse EpiSCs and human ESCs ...44
Chapter 2 Figure 1 ESC-like iPSCs from six diverse inbred mouse strains ...77
Figure 2 Total number of iPSCs derived from each inbred strain ...77
Figure 3 Initial iPSC colony emersion from NOD and WSB are EpiSC-like ...78
Figure 4 Expanded abnormal NOD/LtJ iPSCs ...79
Figure 5 Successful integration of reprogramming vector in NOD iPSCs ...80
Figure 6 Selected NOD iPSCs are karyotypically abnormal ...80
Figure 7 Selected NOD and WSB iPSC colonies differentiate and cannot be expanded ....81
Figure 8 Initial F1 colonies prior to selection ...83
Figure 9 Selected and expanded F1 iPSC colonies ...83
Figure 10 WSB x NOD iPSC colonies grown in EpiSC media are EpiSC-like ...84
Figure 11 129 and NOD F1 hybrids are positive for PECAM1 cell surface expression ...84
Figure 12 WSB x NOD iPSCs are ESC-like on MEFs and EpiSC-like on Gelatin ...85
Figure 13 Treatment methods with chromatin and signal modifying chemicals ...88
Figure 14 NOD iPSCs derived in 2mM VPA resemble NOD untreated colonies ...88
Figure 15 VPA 2mM ...89
Figure 16 Selected NOD iPSCs derived in 2uM VPA ...89
Figure 17 Serum free conditions during reprogramming leads to cell death ...91
Figure 18 2mM VPA + 2i serum free treatment leads to cell death ...92
Figure 19 Emerging iPSC.2iS colonies gradually become ESC-like in culture ...95
Figure 20 iPSC.2iS colonies are ESC-like in culture ...95
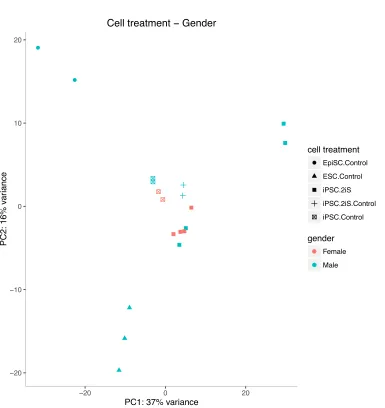
Figure 22 Global gene expression PCA plot of cell treatment and gender for all samples ...99
Figure 23 NOD male iPSCs differ transcriptionally from other male and female iPSCs ....101
Figure 24 iPSCs are more similar to the ESC state than to the EpiSC state ...104
Figure 25 iPSC vs. ESC and iPSC vs. EpiSC comparisons excluding WSB ESC ...106
Figure 26 Difference between 129 iPSCs maintained in 2iS media vs. no 2iS media ...109
Figure 27 Genetic background affects transcriptional difference among iPSCs ...111
Chapter 3 Figure 1 Method behind shortening the 2iS treatment ...153
Figure 2 There is a loss of colony stability and structure once 2iS is removed ...154
Figure 3 Flat and round colonies within the same colony expansion ...155
Figure 4 iPSCs after 2iS removal could not be expanded with trypsin ...156
Figure 5 Increased passage transitions colonies to ESC-like phenotype on MEFs ...156
Figure 6 iPSCs are sensitive to growth on gelatin ...157
Figure 7 WSB x NOD iPSCs are similar to iPSCs derived in short 2iS treatment ...158
Figure 8 Control NOD ESCs and NOD EpiSCs ...159
Figure 9 2iS transitions heterogeneous populations to homogeneous PECAM1 ...160
Figure 10 WSB x NOD iPSCs are heterogeneous cell populations ...161
Figure 11 ESCs and iPSCs derived from permissive strains stain differently ...163
Figure 12 2iS removal transfers homogenous populations to a heterogeneous state ...165
Figure 13 Prediction of single cell separation according to cell state ...168
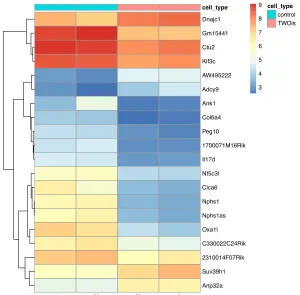
Figure 14 Hierarchical map of the top 200 genes influencing sample similarity ...170
Figure 15 Heatmap of the genes separating the heterogeneous samples ...172
Figure 16 Single cells cluster by presence in/out of 2iS if subgroup is removed ...176
Figure 17 Predicted activity of LIF pathway when cells are grown in 2iS ...181
Figure 18 Majority of cells in heterogeneous populations are PECAM1+ ...183
Figure 19 Repeat of flow sorting experiment ...184
Figure 20 CD40+ cells were not among the captured single cells ...187
Figure 21 Similarly stained iPSCs are more similar than differentially stained iPSCs ...189
Figure 22 PECAM1+ and dual expressing cells are closer to the ESC than EpiSC state ...190
Figure 24 FOXO1 and MYC have higher expression in PECAM1+ cells ...195 Figure 25 Transcriptional differences between Early and Never populations ...196 Figure 26 The error in the Fluidigm protocol explained ...204 Chapter 4
CHAPTER 1 INTRODUCTION Stem Cells
Stem cells have the unique ability to develop into different cell types of the body, divide without limit, and often serve as an internal repair system. These qualities have placed stem cells in an important role for developmental research and medical advances. There are two main categories of stem cells, somatic stem cells and embryonic stem cells.
A somatic stem cell is an adult cell that can indefinitely self renew, but is multipotent and can only differentiate into cell types from the tissue in which it originated. For example, hematopoietic cells are adult stem cells that can differentiate into several types of blood cells including lymphocytes, monocytes, and neutrophils, but cannot differentiate into non-blood cell types such as nerve cells or bone cells. Adult stem cells function throughout the adult body in specified roles. They are found in the gut or bone marrow where they consistently repair or renew damaged cells or in the pancreas and heart where they are utilized under very specific conditions [reviewed in 240]. The biological functionality of adult stem cells has also been used in medicine as exemplified when bone marrow stem cells transplants are used as a treatment for leukemia.
giving rise to the entire organism. This early, crucial developmental time point has imbued embryonic stem cells (ESCs) with key defining features: they are proliferative and self-renewing, but also pluripotent in their ability to develop into any cell type of the three somatic lineages including the germline [reviewed in 241-245]. These qualities make these cells one of the best models to study early development, differentiation, and cell state specialization. Embryonic stem cells can also be used to develop and screen new drugs and hold great potential for regenerative cell-based therapies for disease treatment.
Mouse ESCs were first derived in 1981 and are characterized generally by a
developmentally naïve state [6,7]. This discovery led to derivation of human ESCs in 1998 [8]. Human embryonic stem cells are characterized by a more developmentally primed state, but are still pluripotent. Human embryos used to derive ESCs are donated for research purposes with informed consent from in vitro fertilization attempts. However, research on human ESCs continues to be limited due to ethical concerns. Furthermore, both human ESCs and adult stem cell regenerative medicine is limited by transplant rejection rates caused by histoincompatibilities.
Cellular Reprogramming Early Cellular Reprogramming
cellular reprogramming occurs naturally, when primordial germ cells that are unipotent in vivo become pluripotent upon extraction and growth in culture [38,39]. Furthermore, studies allude to the dynamic state of “committed” cells of the embryo that change when the cells are explanted or exposed to a different microenvironment [reviewed in 15]. Research from Hadorn and Gehring in 1966 and 1967, respectively, show that cells from the imaginal discs of Drosophila melanogaster pupae when serially explanted into the abdomen of an adult fly undergo transdetermination. Cells that were fated to develop into genital structures,
developed instead into leg and head structures and later into wings upon further
transplantation [22,23]. Le Lievre in 1975 demonstrated the concept of cell state plasticity by using an avian model. Neural crest cells from quail were transplanted to chicken, where they adopted new fates and developed into bone, cartilage, and connective tissue guided by their new cellular environment and not their initial location in the quail embryo [24]. All of these studies indicate that cell fate is not permanent and can be altered by the factors in the environment. However, it is the results from two nuclear reprogramming approaches that definitively proved the dynamic nature of cell fate determination.
Single Cell Nuclear Transfer
genes are not permanently inactivated, and that somatic cells retain all the genetic information needed for development.
were retained in a somatic cell and could be activated by unknown reprogramming factors present in the cytoplasm of an earlier cell state, the oocyte.
However, other scientists were still skeptical about the single cell nuclear transfer due to its low efficiency rates and difficulty to replicate in other organisms [15]. The technique was not fully accepted until it was utilized for the successful cloning of Dolly the sheep in 1997 [13]. The successful cloning of the first mammal was attributed to the use of
unfertilized oocytes, and donor cells that were serum deprived in culture, a technique
proposed to induce the cell to exit the cell cycle and alter its chromatin structure. Alterations to this technique led to the successful cloning of mice and ultimately to the cloning of dogs, goats, mules, wildcats, and wolves [14,15]. However, abnormalities including aberrant gene expression in embryos, telomere elongation, obesity in adults, impaired immune system, increased cancer susceptibility, and even death was observed in clones [16-21]. Ultimately, cloning efficiency is dependent on cell type, developmental age, and origin of the donor cell. Despite the contributions of single cell nuclear transfer to the knowledge that cell fate is not fixed, the functional defects seen in clones is an indication of the effect of epigenetics on determining cell state and the success of nuclear reprogramming.
Cell Fusion
Cell fusion is the merging of two cell types to form a single cell, using an external stimulus, typically an electrical pulse. There are two possible outcomes of cell fusion,
species and the single fused cell undergoes cell division an ultimately DNA replication, then a euploid hybrid cell will result. If the cells are not from the same species then the resulting hybrid will typically be aneuploidy [reviewed in 15].
Research on hybrids from different species was limited by chromosome
rearrangements and loss. Heterokaryon research provided the benefit of two separate, intact nuclei that allowed for easy identification of gene products and reprogramming assessment. Early heterokaryon studies showed nuclear swelling and DNA / RNA synthesis, but not the reactivation of silenced genes [reviewed in 15]. In 1983, Blau and colleagues provided the first evidence that previously silent genes could be reactivated in mammalian cells by using heterokaryons formed from the fusion of mouse muscle cells and human amniotic cells. The resultant heterokaryons expressed human muscle proteins, indicating that muscle genes had been activated in non-muscle cells [35].
Cell state is stable once established and is maintained by the continued expression of cell state specific regulators. However, cell state is susceptible to change once exposed to a differing cellular environment containing alternative lineage specific regulators. Together the results from single cell nuclear transfer and cell fusion confirm that the differentiated state of mammalian somatic cells is reversible. Somatic cells retain all the genetic information necessary to revert back to an early embryonic stem cell state and are capable of reactivating previously silenced pluripotency genes. The discovery that cell specialization is not a change in gene content, but rather in gene expression was the guiding principle that lead to the discovery of transcription-based reprogramming.
Transcription Factor Reprogramming
Transcription factor reprogramming is the conversion of one cell state to another by the ectopic expression of lineage specific transcription factors. The concept of transcription factor reprogramming was first demonstrated in 1987 in Drosophila melanogaster larvae, when the ectopic overexpression of the homeotic gene Antennapedia led to the development of legs instead of antenna [25]. Later it was shown that ectopic expression of the Drosophila gene eyeless (also known as Pax6 in mice), a master regulator of a cascade of 2,500 genes, led to the development of functional eyes on the legs, wings, and antennae [26]. In mice transcription factor reprogramming was first demonstrated when ectopic expression of the skeletal muscle factor, muscle helix-loop-helix (MyoD), in fibroblast cells led to the
and T cells into functional macrophages by the overexpression of the myeloid transcription factor, C/EBPα; [28, 29] the conversion of pancreatic acinar cells to insulin-producing beta cells by the overexpression of the pancreatic factors MafA, Pdx1, and Ngn3 [30]; the
conversion of fibroblasts into neurons by the overexpression of Ascl1, Brn2, and Myt1l [31]; and the conversion of fibroblasts into cardiomyocytes by overexpression of the cardiac factors Gata4, Mef2c, and Tbx2 [32]. These early transcription factor experiments
demonstrate that lineage conversion is not limited to cell types within the same lineage [33]. The ectopic overexpression of cell-state specific transcription factors can successfully convert one cell type to another.
Induced Pluripotent Stem Cells Discovery
The discovery using single cell nuclear transfer or cell fusion that a somatic cell can be developmentally reversed to an early embryonic stem cell state and the discovery by early transcription factor experiments that overexpression of lineage-specific master regulators can covert one cell type to another, laid the foundation for the discovery of induced pluripotent stem cells. Induced pluripotent stem cells are differentiated cells that have been converted, induced, or reprogrammed into an embryonic stem cell state by the ectopic overexpression of four embryonic stem cell specific transcription factors, Oct4, Klf4, Sox2, and Myc,
In a classically designed experiment, researchers Yamanaka and Takashi assessed the ability of 24 pluripotency associated candidate genes to convert primarily differentiated mouse embryonic fibroblasts cells to the embryonic stem cell state. The 24 candidate genes were introduced via retroviral transduction into fibroblasts of Fbx15bgeo/bgeo mice and the cells were grown in G418 containing media, an aminoglycoside antibiotic with conferred
resistance to the neomycin gene. If the cells were successfully converted from the fibroblast state to the embryonic stem cell state by the factors, then the embryonic stem cell specific locus Fbx15 containing a β-galactosidase & neomycin fused reporter cassette would become activated, thereby activating the neomycin gene and conferring resistance to the cells. Coexpression of all 24 factors was successful in converting the cells. Using the process of elimination the researchers identified the minimal core set of pluripotency genes needed to successfully reprogram differentiated cells to the embryonic stem cell lineage [1].
Advantages / Benefits
development and cell specialization, but also as an attractive therapeutic tool for personalized cell therapy and disease modeling. Human iPSCs became an attractive alternative to human embryonic stem cells, and as a model for the human condition so did mouse iPSCs.
Mouse iPSCs were used in a proof of concept experiment as a personalized cell therapy tool for the treatment of sickle cell anemia. Sickle cell anemia is caused by a single point mutation in the hemoglobin gene that renders red blood cells nonfunctional due to an abnormal sickle shape. Researchers removed skin cells from a mouse model for sickle cell anemia and reprogrammed the cells into iPSCs. Using gene targeting the point mutation was corrected, the cells were differentiated into blood-forming progenitors, and transplanted back into anemic mice where they produced normal blood cells and cured the disease [41]. In another example, hemophilia A mice transplanted with iPSC-derived progenitor cells cured the hemophilia A condition [42]. This concept can be applied to other human conditions where the genetic mutation is known.
Disease modeling using iPSCs can be used to investigate treatments for diseases with more complex etiologies, such as type I diabetes, Alzheimer’s disease, and Parkinson’s disease. The goal of disease modeling is to create a petri dish model of the disease to investigate novel drugs that could alleviate symptoms or prevent the loss of essential cells, such as insulin-producing B cells in diabetes patients or motor neurons in patients suffering from amyotrophic lateral sclerosis (ALS) and spinal muscular atrophy (SMA). To
to witness the progression of the disease to more advanced stages as the iPSCs will also have to undergo the same progression in their differentiation process. This allows researchers to gain insight into the earliest stages of the disease [reviewed in 33]. Several labs have already derived iPSCs from patient’s battling Huntington’s disease, Parkinson’s disease, ALS, juvenile diabetes, muscular dystrophy, Fanconi anemia, and Down syndrome [43-46]. Some labs have also replicated disease conditions within a petri dish for SMA, familial
dysautonomia, and LEOPARD syndrome and even identified experimental drugs to alleviate disease symptoms in culture [47-49].
On September 12, 2014 a 70-year-old Japanese woman suffering from severe age-related macular degeneration received the first long awaited and highly anticipated patient specific iPSC treatment. The patient suffered from severe age related macular degeneration, which is caused by the invasion of blood vessels into the retina, resulting in the destruction of the retinal pigment epithelium, a layer of cells that support photoreceptors necessary for vision. The iPSC derived retinal cells were implanted during a two-hour surgery. While the implantation of the cells was not anticipated to improve the patient’s vision, it was expected to stop the retina from deteriorating. As the research on iPSCs began to boom, so did
bleeding or other complications during the surgery and will be monitored for a year [239]. If the efforts of Masayo Takahashi’s team are successful it could usher in an era of new
approval for iPSC therapeutics. If it is unsuccessful, it could set the field back and make approval for clinical use harder to acquire.
iPSC & ESC Equivalency
The possibility of iPSCs to be used as treatments and as scientific research tools raises the valid question of their equivalency to true embryonic stem cells. The
first-generation mouse iPSCs created by Yamanaka and Takashi fulfilled some of the criteria of pluripotency by generating teratomas and contributing to the developing embryo upon blastocyst injection. However, they also expressed lower levels of key pluripotency genes such as Oct4 when compared to true embryonic stem cells and failed to contribute to the germline of chimeras [1]. This suggests that the early iPSCs, selected with Fbx15
reactivation were only partially reprogrammed [33]. Later studies found that the selection of iPSCs based on the reactivation of pluripotency genes, such as Nanog or Oct4 were more successful in identifying iPSCs that closely resembled ESCs both molecularly and
functionally [50-52].
degree of similarity [50-52,55]. However, some studies reported a difference in the potential of iPSCs to function as truly naïve stem cells. One study reported that there was an increased likelihood of iPSCs derived from neural cells to form tumors after transplantation into mouse brains. Note that the influence of residual transgene expression was not excluded from this study and could be the likely cause of the finding for increased propensity to form
tumorigenic cells. Another study reported that human iPSCs had more variable and reduced neuronal potential [56]. A larger study identified global and gene specific expression differences in DNA methylation [57-59] and in mRNA and miRNA expression for both mouse and human iPSCs when compared to ESCs [60,61]. Note that again the influence of residual transgene expression [1,62] and genetic background [63], which can impact pluripotency, were not accounted for in these studies [33].
A later study compared the global mRNA and miRNA expression patterns of genetically matched mouse ESCs and iPSCs. There were no consistent differences in gene expression with the exception of the maternally expressed Dlk-Dio3 cluster [64]. In true embryonic stem cells maternally expressed genes within the Dlk-Dio3 cluster, such as the Gtl2 gene in mice, are activated. In iPSCs derived from fibroblasts, blood cells, and keratinocytes maternally expressed genes within the cluster were aberrantly silenced. Induced pluripotent stem cells with aberrant gene silencing of the maternal cluster still met the criteria of pluripotency by forming teratomas and contributing to low-grade chimeras. However, iPSCs with ESC-like expression of the Dlk-Dio3 cluster not only formed
fibroblasts have been found to have normal Dlk-Dio3 cluster expression, likely due to higher expression of the cluster in fibroblast cells. The finding that there were no consistent
differences in overall gene expression suggests that previously reported transcriptional differences might be due to other experimental variables [reviewed in 33].
The concept of an iPSC retaining an epigenetic memory of its previous cell state supports the idea of residual gene expression differences among iPSCs limiting their
To some extent, iPSC equivalency and epigenetic memory may be influenced by species-specific differences. An example of this can be observed with the epigenetically controlled X chromosome inactivation in females. Female fibroblast populations are mosaic in that 50% of cells carry an active maternal X chromosome and an inactive paternal X chromosome, whereas the other 50% of the population carry the exact opposite [50]. Human iPSCs derived from female fibroblasts do not reactivate the silenced X chromosome even upon differentiation [68]. In contrast, mouse iPSCs derived from female fibroblasts do reactivate the silenced X chromosome and undergo normal random X inactivation upon differentiation. This suggests that female human iPSCs also retain an epigenetic memory of their previous cell type. However unlike mouse iPSCs, the epigenetic memory of female human iPSCs does not become alleviated with increase passage [reviewed in 33]. Methods of Reprogramming
In the original reprogramming experiments, the four factors (OKSM) were transduced into the genome using four separate retroviral vectors, one for each factor [1]. The retroviral vectors integrated randomly into the genome where they were constitutively expressed. Retroviral transgenes are usually silenced toward the end of the reprogramming process [69] due to activation of DNA [70] and histone methyltransferases [71]. However the use of retroviral vectors leads to a dependency of cells on exogenous factor expression often
potential [1] and lead to tumorgenesis in chimeras [51]. Retroviral reprogramming also has a lower efficiency due to the fact that retroviruses can only infect mitotically active cells.
Lentiviral reprogramming on the other hand has higher efficiency due to the fact that lentiviruses, a subtype of retrovirus, can infect both non-dividing and actively dividing cell types. Reprogramming efficiency is also enhanced by the use of lentiviral polycistronic cassettes encoding all four factors as opposed to infecting cells with four separate retroviruses [73,74]. However, the issue of constitutive expression from a randomly integrated virus is enhanced with lentiviral vectors due to their inability to be efficiently silenced in pluripotent cells [75,76]. One solution for this is the use of inducible lentiviral vectors that can be controlled by the presence or absence of the drug doxycycline in the media [69,75]. Another solution is the use of flanking loxP sites that can excise the reprogramming cassette from the genome by transient expression of Cre recombinase [77,78]. An alternative transgene free equivalent of integrating, but excisable lentiviral reprogramming is the use of piggyBack transposons that can be introduced into the genome and removed by transient expression of a transposase [79, 80].
factors [reviewed in 33]. Purified recombinant proteins supplemented with the histone deactylase (HDAC) inhibitor VPA [85], whole cell extracts from ESCs [86], and genetically engineered HEK293 cells [87] have also been used, albeit very inefficiently, as a DNA-free method of reprogramming. The addition of modified RNA molecules encoding the
reprogramming factors into somatic cells has also been used more successfully as an alternative DNA-free approach [88].
Due to low efficiency and infrequent use of alternative methods the remainder of this dissertation will focus on the lentiviral approach to reprogramming.
Functions of the Four Factors
As previously mentioned, transcription factor mediated reprogramming is controlled by a set of four embryonic stem cell transcription factors – OCT4, SOX2, KLF4, and MYC, collectively referred to as OKSM. Each factor plays a critical role in reprogramming. OCT4, SOX2, and KLF4 are part of a key pluripotency network, which includes a fifth ancillary member NANOG [89]. Together this key pluripotency network is responsible for repressing genes associated with the somatic program and activating genes associated with the
pluripotency state [90,91,92].
polymerase to pause in response to a perceived error [95, 96]. The RNA polymerase then returns to the beginning of the transcript to transcribe the same gene again. This not only leads to the amplification of target pluripotency genes, but also reduces rate-limiting constraints for tumor cell growth making Myc a potent oncogene. Consequently, MYC expression also stimulates cell proliferation and induces a metabolic switch from an oxidative to a glycolytic state resembling ESCs [93, 72]. Early expression of MYC and to some extent KLF4 in fibroblasts increases reprogramming efficiencies and speed. Activation of KLF4 target genes have been found during both early and late reprogramming stages, indicating that KLF4 functions similarly to MYC in facilitating early reprogramming events [93].
KLF4 also functions similarly to OCT4 and SOX2 later during reprogramming to activate genes necessary to maintain pluripotency [97]. The presence of MYC on promoters of active genes and on microRNAs suggests that it may also play a later role in solidifying pluripotency [98, 99, 100]. MYC is the most dispensable of the four factors because it does not directly enhance pluripotency. Removal of MYC does not affect the ability to form iPSCs, but it does reduce efficiency and speed [101, 102]. One reason for MYC’s
dispensability is that it is already expressed at low levels in many somatic cells. Research on the reprogramming of neural stem cells to iPSCs with only OCT4, suggests that factors that are endogenously expressed within the somatic cell may be excluded from the
reprogramming cocktail [103].
and LIN28 [104], which represses Let-7 miRNAs that negatively regulate Myc translation [105]. Therefore, repression of Let-7 miRNAs leads to the natural upregulation of Myc [106,107]. SOX2 and KLF4 can be replaced with the functionally similar proteins, SOX1 and KLF2, which may indicate the recognition of similar DNA-binding motifs [101].
Alternatively, proteins with completely different functions have also been found as substitutes. Examples of this are the substitution of KLF4 with ESRRB [108] and the substitution of OCT4 with the orphan nuclear receptor NR5A2 [109]. These results indicate remarkable redundancy in the functionality of gene networks and the variety of pathways that can be taken to achieve pluripotency.
Regardless of these substitutions, OKSM remains the most successful and commonly used combination of factors to induce pluripotency. As a result the remainder of this
dissertation will focus on the result of reprogramming when these factors are utilized. Models of Reprogramming
The reprogramming process in most somatic cells is extremely inefficient, with success rates ranging from 0.001% to 1% at best [reviewed in 33]. Two competing
the reason for low reprogramming efficiency is that only a few cells will successfully pass through all the epigenetic roadblocks of reprogramming [reviewed in 33].
Individually both hypotheses can be refuted by experimental findings. The elite model is undermined by the success of reprogramming multiple cell types including fully differentiated B and T lymphocytes [111,112] in addition to pancreatic beta cells [113]. Furthermore, when given a sufficient amount of time ranging from several weeks to several months, all B cells and monocytes expressing the four reprogramming factors formed iPSCs [114]. This suggests that with continual cell proliferation and enough time, even rare cells within a homogenous population will undergo the changes necessary for reprogramming. The limit of the stochastic model is observed when the speed of reprogramming for cells types from terminally and primarily differentiated cell lineages is analyzed. Hematopoietic stem and progenitor cells, which already express some pluripotency factors, undergo shorter reprogramming times than mature lymphocytes and myeloid cells regardless of the
proliferative state of cells during factor introduction [112]. This is expected, considering that cells that already express some pluripotency factors will have to pass through fewer somatic gene-silencing events than a fully differentiated cell.
A better model would be a stochastic model that integrates elite or deterministic components [33]. Certain cells within a given population are predisposed to be more easily reprogrammed, but all cells have to progress through the same random processes of
constantly undergoing subtle shifts between different cell states. There may also be subtle differences in the cell culture environment as well as influential, but undetermined
relationships, between neighboring cells. It is almost impossible to control all the variables homogenously. This is particularly exacerbated with fibroblasts that may contain a mix of different cell types. This is supported by the finding that subpopulations of fibroblasts are more successful at giving rise to iPSCs than the overall bulk population [115]. Furthermore, low expression of the tumor suppressor gene Arf is used as an indicator of which fibroblast cells will be more susceptible to reprogramming [116]. This suggests that there may be intrinsic cellular differences within a given population of cells not determined by a differentiation hierarchy.
It is difficult to predict which cell types will reprogram, but the rate of
different for each cell type, laboratory, and method [117]. As discussed previously, there are many routes that lead to cellular reprogramming. Here I will discuses what is known to occur during epigenetic reprogramming using a four factor based method.
Epigenetic Reprogramming Epigenetic Model
Most of what is known about epigenetic reprogramming is based on the
experimentally deduced role of the OKSM factors in targeting specific genes to reshape the epigenetic landscape. Conrad Waddington coined the term epigenetics in 1942, which was defined more extensively in terms of the epigenetic landscape model in 1957 [4,5]. The epigenetic landscape model illustrates the developmental pathways a cell can take toward differentiation. It depicts a ball rolling down a hill that is sloped with ridges. The ball at the top of the hill represents a cell in a fully naïve state. This is known as the “competent” potential of the cell to be pushed through induction (signal) down a developmental pathway. As the ball rolls down the hill it begins to differentiate eventually falling into one of the available valleys or pathways. The ridges that separate each valley define the regulation limits of each pathway. For example, once the ball has reached the lowest point in the valley, the ridges prevent it from rolling into another valley. Likewise, once a cell has fully
switch cellular fates. Not usually shown is the underside of Waddington’s model, which illustrates the landscape being connected by strings to a matrix of pegs. The pegs represent genes and the strings represent gene expression and signaling networks that pull at the landscape above causing it to change shape in response to the cellular environment. This symbolizes the ability of genes to regulate the epigenetic landscape [5] Taken together in light of iPSC derivation, the overexpression of some genes can change the tension of the strings resulting in the collapse of ridges, ultimately changing the epigenetic landscape and allowing a shift in cell fate.
OKSM Factors
The OKSM factors reshape the epigenetic landscape by binding directly to target sites, by binding indirectly to other factors that activate genes necessary in reprogramming, or by recruiting other proteins that change chromatin conformation. Because the OKSM factors are expressed in the embryonic cell state, many of its target genes are inaccessible in somatic cells due to the chromatin conformation. Therefore, OKSM targets can be classified into three classes based on chromatin state. The OKSM factors first bind completely
reprogramming events to occur and is vital to extinguishing gene expression of the somatic program [reviewed in 89].
The second group of targets is comprised mostly of distal regulatory elements in a somewhat open chromatin state. The activation of these genes requires chromatin remodeling [118]. Similar to permissive enhancers, these genes express the H3K4me1 mark, nucleosome depletion, and DNaseI hypersensitivity. Permissive enhancers allow for the binding of transcription factors before their associated promoters [119]. The Myod1 locus is a classic example of a permissive enhancer. OCT4 binds to the enhancer region of the Myod1 locus, which triggers crosstalk with the Myod1 promoter and leads to the establishment of a poised chromatin state [119]. DNaseI resistant loci may also fall into to this second category of OKSM targets. In such cases, targets of OKSM function as pioneering factors that bind to closed chromatin and initiate chromatin remodeling. For example, although early MYC binding establishes an open chromatin state, some genes such as Sal4, act as an early pluripotency gene, are DNaseI resistant and cannot bind to MYC. In this second step of reprogramming, the remaining OKS factors bind these resistant loci to facilitate the binding of MYC and establish a more open chromatin state [118].
The third group of OKSM targets is comprised of completely closed or
expressed in this third class with completely closed chromatin states require the most chromatin remodeling.
Histone Modifying Enzymes
Histone modifying enzymes play an important role in the regulation of chromatin state and in reprogramming. They function as co-activators or co-repressors of OKSM [reviewed in 89]. There are two categories of histone modifying enzymes, writers and
erasers. Histone writers are histone methyltransferases (HMTs) and histone acetyltransferases (HATs) that add methyl or acetyl groups to the histone tails. Histone erasers are histone demethylases (HDMs) and histone deacetylases (HDACs) that remove methyl or acetyl groups from histone tails.
HDMs play a critical role in the removal of repressive marks to increase
reprogramming efficiency. For example, the addition of vitamin C during reprogramming activates the HDMs, JHDM1A and JHDM1B that enhance reprogramming through H3K36me2/3 demethylation in mouse embryonic fibroblasts [125]. Activation of the JHDM1A and JHDM1B H3K35 HDMs and the suppression of the JMJD3 HDM promote suppression of the Ink4/Arf locus during intermediate to late stages of reprogramming [126]. The Ink4/Arf locus stimulates cell senescence and its knockdown has been found to greatly increase reprogramming efficiency [116, 130, 131, 132, 133].
JMJD3 also functions in a HDM independent manner to inhibit reprogramming by targeting the methyl-lysine effector protein PHF20 for ubiquitination. PHF20 interacts with WDR5 to reactive endogenous Oct4. MEFs with no PH20 expression are not converted to fully reprogrammed iPSCs. It is therefore, critical to downregulate Jmjd3 to both suppress the INK4A/ARF pathway and to increase PHF20 expression [126]. Together the above results demonstrate the critical role that histone modifiers play during reprogramming by either maintaining cells in a somatic state or by removing the somatic state to assist the reprogramming factors in the establishment of pluripotency.
OCT4 has a unique linker domain that is not shared with other POU domain family members. This linker domain directly associates with chromatin remodelers to overcome epigenetic barriers to reprogramming and remodel chromatin. The removal of this essential linker domain from OCT4 completely halts reprogramming [129], demonstrating both its importance and the significance of remodeling chromatin in the reprogramming process. Chromatin Architecture
The OKSM factors act as chromatin organizers that rearrange chromatin architecture during reprogramming to establish pluripotency [reviewed in 89]. Chromatin architecture is defined as the both the presence of histone variants and the position and density of
nucleosomes [134]. Chromatin architecture is altered though chromatin remodeling
barrier to reprogramming by maintaining the heterochromatic state. Knockdown of the NURD complex increases iPSC generation [137,138].
In addition to local chromatin structure, OCT4 has also been implicated in 3D
chromatin architecture by participating in long-range chromatin interactions in cells poised to become iPSCs during reprogramming in both mice and humans [139,140]. The idea that some cells are poised to become iPSCs harkens back to the before mentioned elite cell theory of reprogramming. In other studies KLF1, a functionally similar protein to KLF4, has also been implicated in addition to OCT4 in participating in long-range chromatin interactions [141,142]. Together these results suggest that the OKSM factors do not merely activate or deactivate genes, but also function to reorganize chromatin during the reprogramming process.
DNA methylation
DNA methylation occurs last during reprogramming, after histone and chromatin changes have already occurred and is the most stable epigenetic modification [93, 144, 89]. The demethylation of pluripotency genes is crucial to forming ESC-like iPSCs. Incomplete DNA methylation during the reprogramming process has been shown to lead to retained epigenetic memory of previous cell fate [65, 66]. DNA methylation is established by DNMT3A and DNMT3B and maintained by DNMT1 [reviewed in 143].
It also supports the idea that in mice, the silencing of lineage specific genes is not achieved through DNA demethylation, but rather through alternative chromatin based mechanisms such as the activation of the polycomb repressive complex that establishes repressive H3K27me marks on somatic genes [121,89].
Although DNA methylation occurs last, it is not dispensable to reprogramming. Incomplete DNA demethylation leads to retained epigenetic memory and partially
reprogrammed iPSCs [65,66]. There are two mechanisms of DNA demethylation, passive and active [reviewed in 143]. Passive DNA demethylation occurs when the newly
synthesized DNA strand is not methylated by DNMT1 during several replication rounds. Passive DNA demethylation plays a supportive role during reprogramming by facilitating replication dependent DNA methylation. In fact, the downregulation of DNMT1 results in the transition of intermediate iPSCs to fully reprogrammed iPSCs [55].
Active DNA demethylation plays a more critical role in reprogramming [reviewed in 89]. Active DNA methylation is replication independent and occurs through the direct removal of methyl groups in three steps. Step one; the enzyme TET hydroxylates 5-methylcytosine (5mC) to form hydroxy5-methylcytosine (5hmC). Step two; the enzyme
activation-induced deaminase (AID) deaminates 5hmC to form 5-methyluracil (5-hmU). This triggers the cells repair mechanisms. In step three; the TDG and SMUGI base excision repair glycosylases replace 5hmU with cytosine [reviewed in 143].
Esrrb, priming them for demethylation and subsequent activation [146]. Furthermore, NANOG and TET1/TET2 interact directly to co-occupy other pluripotency targets [147] In support of this cooperation, the overexpression of NANOG with either TET enzyme
enhances iPSC formation, where as the loss of NANOG and TET2 stops iPSC formation [146-148]. Additionally, TET1 has been found to compensate for loss of exogenous OCT4 expression during reprogramming [149]. This suggests that tet1 may play a similar role in the activation of OCT4 target pluripotency genes.
OKSM accessibility to target chromatin regions may influence the efficiency of DNA demethylation [118]. Broad H3K9me3 domains that overlap with closed chromatin or
inaccessible OKSM often characterize genomic regions that fail to undergo DNA
demethylation in human iPSCs [150]. This also harkens back to the importance of HDMs or HMTs in removing epigenetic marks directly on the chromatin or indirectly during DNA methylation. Together these results implicate the effect of the OKSM factors on epigenetic reprogramming and suggest that their function may be the most influential factor in forming ESC-like iPSCs.
The Mouse as a Model
Brief History & Practicality of Mouse as a Model
when William E. Castle, one of the founding fathers of mammalian genetics and the first director of the Bussey Institute of Experimental Biology at Harvard, legitimized the use of mice in biomedical research. Mice quickly became the mammal of choice to model genetics because of its small size, resistance to infection, larger litter size, and rapid generation time. Mice also became favored because of mutations that produced an interesting natural variation of coat colors and behaviors. Together, researchers and mouse pet “fancy” breeders such as the mother of mouse genetics, Abbie E. C. Lanthrop, worked to create the earliest mouse breeding experiments [reviewed in 152].
Today, the mouse is still regarded as one of the most attractive mammalian models of the human condition. Mouse and humans share a high degree of similarity in their anatomy, physiology, and genetics. Ninety-nine percent of human genes have orthologous mouse genes, making the mouse an applicable model to investigate human development, evolution, and disease [154]. Mice and the humans share genes for hypertension and atherosclerosis and can be used as a model to predict genetic risk [reviewed in153]. Additionally, there are many naturally occurring spontaneous mutations in mice that mimic disease conditions.
Mice are easily manipulated, cost effective, require less space and time, and have established genomic resources, making the mouse a practical model choice. Mice are small and can be easily housed several to a cage in specialized facilities. They have a short
through the external introduction of genes to produce iPSCs has also been made. Lastly, embryos, sperm, and cell can now be effectively cryopreserved. In addition to technical manipulation advances, advancing in breeding experimentation has produced vital resources for gene vs. environmental discovery [reviewed in 152, 153] The creation of recombinant inbred strains and diversity-outbred strains are just two examples of the many manipulations that can be made to the mouse model.
The natural variation in coat color that was appreciated and used by early researchers was only one aspect of mouse variation. Early breeding experiments lead to the production of inbred mouse strains, many of which serve as useful models for various human conditions and diseases. An inbred strain is a strain that has undergone more than 20 generations of brother sister mating, resulting in a strain of mice that is homozygous at all genetic loci except for spontaneous mutations [155,156]. The creation of inbred strains has provided a resource to model complex diseases involving multiple gene interactions and has also allowed for the creation of several mouse models that are reflective of a particular human trait. One such example is the non-obese diabetic mouse, NOD, which is used to model human type II diabetes (236). Today the usefulness of inbred strains has expanded to the creation of recombinant inbred strains, such as the Collaborative Cross (CC) population, which offers a genetically defined and amenable model system to map and examine
CC captures approximately 90% of the genetic variation of laboratory mice and is comparable to the genetic variation found in human populations [246]. This population structure places the CC in the ideal position to assign genetic causality and to study the multifarious biological networks underlying mammalian traits. However, the CC cannot capture the level of recombination found in populations as easily as diversity-outbred strains. Therefore, the Diversity Outbred (DO) mouse population, a diverse heterogeneous
population of mice generated using the eight parental strains of the collaborative cross, was created to capture increased levels of genetic recombination [151].
Mouse Embryonic Development
The mouse is an advantageous model to study biological traits, such as the diverse cellular state outcomes of iPSC reprogramming. The mouse is the only species from which both naïve and developmentally primed pluripotent cells can be derived without additives. These cell types can be found naturally at various stages of mouse embryonic development.
Mouse development proceeds through several distinct states of pluripotency (Fig. 1). There are two segregation events in the developing embryo that separate the extra-embryonic trophectoderm and primitive endoderm lineages from the pluripotent primitive ectoderm or epiblast lineage, from which all cells of the adult organism will arise [148,159]. The
trophectoderm lineage is the first to separate and can be found on the outside of the early pre-implantation blastocyst at 3.5 dpc. At this developmental time point cells that will form the primitive endoderm and the primitive ectoderm are both found within the inner cell mass (ICM) of the blastocyst. The ICM is characterized by transcriptional heterogeneity at the single cell level; with cells expressing the pluripotency factor NANOG and the
developmentally primed factor GATA6 in a random “salt and pepper” fashion [157]. This is the same developmental time point that ESCs are taken from the inner cell mass and derived. Therefore, it is not unreasonable that these cells maintain transcriptional heterogeneity in culture. Transcriptional homogeneity occurs after the separation of the primitive endoderm from the primitive ectoderm at the mature blastocyst stage found at implantation 4.5 dpc [158]. This ground state of pluripotency is characterized by the expression of key
methylation at pluripotency genes, that prevent its integration into developing blastocyst chimeras and prime it to differentiate into the cell lineages of the adult organism [159,161].
Cell$Lineage$Color$Key$
Trophectoderm$$
Primi6ve$Endoderm$$ Epiblast$/$Primi6ve$Ectoderm$$ Primordial$Germ$Cells$$
Anterior$Visceral$Endoderm$
5.5#dpc# Mature#Epiblast#
Naïve#State#of#
Pluripotency## Ground#State#of#Pluripotency## Primed#State#of#Pluripotency## ICM#
3.5#dpc# Pre@implantaAon# Early#Blastocyst#
4.5#dpc# ImplantaAon# Mature# Blastocyst#
5.5#dpc# Post@ImplantaAon# Early#Epiblast#
ESCs# 2i# EpiSCs#
In#Vivo#Stage##
Pluripotency# ClassificaAon#
In#Vitro#Model#
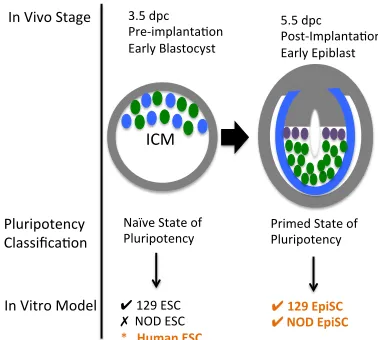
Figure 1: Pluripotency and mouse development
ESCs are derived from the inner cell mass (ICM) of the pre-implantation epiblast found in the 3.5 days post conception (dpc). Three major lineages characterize early development, the trophectoderm, the primitive ectoderm (epiblast), and the primitive endoderm. At this stage the first lineage separation from the trophectoderm has occurred but the second lineage separation between the primitive ectoderm and the primitive ectoderm has yet to occur. The ICM of the early 3.5 dpc blastocyst is heterogeneous for NANOG expression as well as other transcription factors. Naturally ESCs derived from the ICM at this stage are also
heterogeneous for a number of transcription factors. This heterogeneity resolves at the 4.5 dpc blastocyst stage following the final lineage separation. The use of 2i culture conditions has been used to maintain ESCs in a transcriptionally homogenous state of ground
Non-Permissive vs. Permissive
The embryonic stem cell state is an in vitro singularity, as ESC immortality only occurs within a lab dish. In fact, some scientists argue that ESCs grown in culture resemble epiblasts [160]. This is true for the majority of species except for select strains of mice, in particular the 129/SvImJ strain that are permissive for naïve embryonic stem cell formation [188]. The term non-permissive refers to the inability of some genetic backgrounds to derive embryonic stem cells from the inner cell mass under the same culture conditions used for the permissive 129/SvImJ mouse strain.
In other species, such as humans, cells isolated from the inner cell mass of the blastocyst more closely resemble both morphologically and molecularly the mouse epiblast stem cell stage of mouse development than the embryonic stem cell stage of mouse
development (Fig. 2) [90, 162,163,169,170]. These putative embryonic stem cells are
Naïve&State&of&
Pluripotency&& Primed&State&of&Pluripotency&&
ICM&
3.5&dpc& Pre<implanta=on& Early&Blastocyst&
5.5&dpc& Post<Implanta=on& Early&Epiblast&
✔129$EpiSC$ ✔$NOD$EpiSC$ &
In&Vivo&Stage&&
Pluripotency& Classifica=on&
In&Vitro&Model& ✔ 129&ESC& ✗&&NOD&ESC& *$$$Human$ESC$
Figure 2: Difference between permissive and non-permissive strains
ESCs can be derived from the inner cell mass of the 3.5 pre-implantation early blastocyst of the permissive 129/SvImJ mouse strain. ESCs cannot be derived from the inner cell mass of the 3.5 dpc pre-implantation blastocyst under standard conditions from the NOD background. The earliest developmental time point that stem cells can be readily derived from the NOD background is the post implantation early epiblast. These stem cells are termed epiblast stem cells (EpiSCs) and require different culture conditions than ESCs. Human ESCs are derived from the inner cell mass of the pre-implantation blastocyst, but are morphologically and molecularly closer to the mouse EpiSC stage than to the mouse ESC stage. Human EpiSCs have not been derived from the post implantation epiblast for ethical reasons.
method to derive ESC-like iPSCs from non-permissive strains. Research into both permissive and non-permissive mouse strains as well as the ESC and EpiSC states may reveal insights into the genetic mechanisms underlying the regulation of these pluripotency states and cellular differentiation.
✗ ✗
✔ ✗
✔ ✔
✗ ✔
PWK/PhJ WSB/EiJ 129S1/SvImJ A/J C57BL/6J NOD/LtJ NZO/H1LtJ
✔
✔ ✗
✗ ✗
✗
Is#the#Ability#to#Form#ESC#a#Derived#
Characteris8c#in#Mice?#
✗
✗ ✗
CAST/EiJ
✗ ✗
Figure 3: The mouse embryonic stem cell state is a derived state
ESCs vs. EpiSCs in vitro
A look at mouse embryogenesis reveals that ESCs and EpiSCs cells are not the same. In vivo, both ESCs and EpiSCs have the potential to give rise to all cell types, but they occur at two distinct developmental time points. The affect of these developmental time points is seen in the characteristics and developmental potential of these cells in vitro.
In vitro, mouse ESCs, are characterized by a developmentally naïve or ground state that allows for easier manipulation in culture and greater developmental potential. Mouse ESCs are characterized by transcriptional marks, such as key pluripotency genes including Oct4, Sox2, and Nanog as well as epigenetic marks, such as pre X-inactivation that maintain the cell in an undifferentiated state [162,161,184]. The undifferentiated state allows mouse ECSs to contribute to all somatic lineages including the germline upon chimera formation.
as grafted EpiSCs in ectopic sites are capable of forming teratomas [162]. Morphologically ESCs are characterized by dome shaped three-dimensional colonies that can be passaged through treatment with trypsin. However, EpiSCs are characterized by flattened colonies that grow poorly following initial treatment with trypsin [162,183].
STAT3$
MYC$ KLF4$NANOG$
β;CATENIN$
SHP2$
RAS$
RAF$
MEK$
ERK$ AKT$
TBX3$ PI3K$
GSK3β$ GSK3β$
(Canonical)$$
WNT$ LIF$Signaling$
SOX2$ OCT4$
(Autocrine)$$ FGF4$ BMP4$
SMAD$1/5/8$
ID$
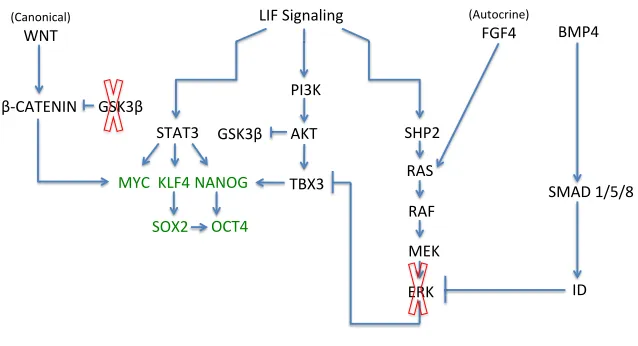
Figure$complied$with$informaUon$from$sources:$185,$189,$191,$247;250!
Figure 4: The LIF pathway activates pluripotency and differentiation pathways The LIF receptor stimulates the STAT3 pathway, which results in MYC, KLF4, and NANOG expression. KLF4 expression stimulates SOX2, which in addition to NANOG activates OCT4. The core pluripotency network is shown in green. MYC stimulates WNT activation, which results in the accumulation of β-CATENIN and further stimulates MYC protein production. The GSK3B inhibitor (red X) prevents the degradation of β-CATENIN by GSK3B, resulting in expression of the canonical WNT pathway and its target pluripotency genes. The PI3K pathway is stimulated by LIF and results in TBX3 expression, which
regulates NANOG. LIF also activates the SHP2/MAPK pathway, which promotes ERK production and cellular differentiation. OCT4 and SOX2 also activate FGF4, resulting ERK production. The BMP4 signally pathway, stimulated by serum-based media, produces inhibitor of differentiation proteins that block ERK production. In 2i-based media, the MEK inhibitor (red X) inhibits the ERK differentiation pathway.
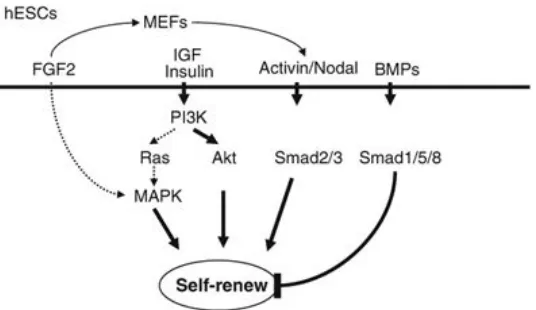
Figure'from'source'251'''
FGF2'&'Ac3vin'Signaling'
Figure 5: FGF2 and ACTIVIN maintain mouse EpiSCs and human ESCs
FGF2 induces TGFβ1/ACTIVIN/NODAL secretion from supporting feeder cells. FGF2 also directly activates TGFβ1/ACTIVIN/NODAL in human ESCs and mouse EpiSCs.
ACTIVIN/NODAL activates SMAD2/3 proteins, which up-regulate the key pluripotency genes Nanog and Oct3/4. Exogenous FGF2 also causes feeder cells to secrete insulin-like growth factor 2 (IGF) that initiate the PI3K pathway and further supports self-renewal.
Embryonic Stem Cell Heterogeneity
At the single cell level embryonic stem cells derived in conventional serum/LIF conditions, homogenously express OCT4 and SOX2, but are heterogeneous for a number of other pluripotency factors including, NANOG, ZFP42 (REX1), Brachyury (T), and DPPA3 (STELLA) [193-200].
markers NANOG and REX1 at high levels, while DPPA3 negative ESCs more closely resemble the developmentally primed EpiSC state and express FGF5 and GBX2 at
intermediate levels between DPPA3 positive cells an epiblast stem cells. DPPA3 positive and negative ESCs also have distinct differentiation potentials, with DPPA3 negative cells being biased to differentiate into somatic and trophectoderm lineages, and DPPA3 positive ESCs biased to differentiate into embryoid bodies [194].
Variable expression of NANOG also confers distinct probabilities of cell renewal and differentiation potential. It has been implicated as one of the key ancillary transcription factors of the pluripotency network [91,94,202,203]. In fact NANOG seems to be a critical indication of ground state pluripotency and cell competency. Cells that lack NANOG have a higher propensity for differentiation and are less likely to integrate into the developing embryo during chimera formation. Approximately 80% of individual embryonic stem cells express NANOG and 10 – 20% of cells do not [193]. Many pluripotency genes, some which are targets of NANOG, continue to be expressed even in the absence of NANOG,
demonstrating that it is not an absolute requirement of pluripotency [160,193]. However, it is critical to preventing counteractive differentiation signals necessary for maintaining cells in an undifferentiated state as cell that lack NANOG expression have an increased tendency to differentiate [193,204]. In fact, forced expression of NANOG prevents ESC differentiation and supports cell renewal even in the presence of FGF/ERK differentiation signals [193,188].
for the balance between self-renewal and the capacity for differentiation. Indeed, the balance of expression is restored after individual cells are isolated and cultured, indicating that transcriptional heterogeneity is a regulated natural occurrence [193,194,205].
Transcriptional heterogeneity occurs in vivo as well and is essential as the ICM or preimplantation epiblast will differentiate into the post implantation epiblast. In vivo the cells of the inner cell mass express both the developmentally naïve marker NANOG and the developmentally primed marker, and GATA6 in a random, but mutually exclusive
heterogeneous manner [157]. It is hypothesized that single cell heterogeneity is an important quality of stem cells populations that allow cells the capacity to differentiate, while still remaining in an pluripotent cell state until subsequent differentiation signals as development proceeds [205,206].
Others argue that conventional ESCs derived are serum/LIF conditions are transcriptionally heterogeneous at the single cell level [193-200, 220-224] because of conflicting signaling pathways activated by LIF and by conflicting undefined signals found in serum based media. LIF is a key molecule used in ESC derivation. LIF signaling
differentiation signals by stimulating inhibitor of differentiation proteins [188]. However, there other numerous undefined, conflicting signaling pathways activated in serum [159] and by OCT4 and SOX2 pluripotency factors [160,207] that are postulated to create a
transcriptional battleground of pluripotency and differentiation expression signals.
The use of two inhibitors in culture, the GSK3B inhibitor and the MEK inhibitor, in serum free conditions commonly referred to as 2i/LIF has been found to transition ESCs to a transcriptionally homogenous landscape similar to the ground state of pluripotency [159, 212, 208, 215-219] that is postulated to occur in vivo in the 4.5 dpc mature blastocyst [159, 157, 214]. The GSK3B inhibitor prevents degradation of beta-catenin thereby enhancing self-renewal and stabilizing ESC propagation by engaging with and enhancing the production of key pluripotency factors [209-213]. The MEK inhibitor blocks activation of differentiation signaling pathways to maintain cells in an undifferentiated state [160,209] (Fig 4).