Improvement of Olefin Metathesis Efficiency
through Understanding Catalyst Stability
Thesis by
Soon Hyeok Hong
In Partial Fulfillment of the Requirements for the Degree of
Doctor of Philosophy
California Institute of Technology Pasadena, California
2007
ii
iii
iv
Acknowledgements
I deeply appreciate many people for their assistance and contributions to the work in this dissertation and for helping me enjoy life as a graduate student.
I want to begin by acknowledging Professor Bob Grubbs for being a wonderful advisor. He has been very patient with me and provided a unique environment to do chemistry. The freedom which Bob provided allowed me to pursue some interesting challenges for myself and participate in research projects beyond a mere student. Yet at the same time, he guided me to approach problems in the right direction at some critical points. I am very honored to have had the great opportunity to work in his group.
I thank my committee member, Professors John Bercaw, David Tirrell, Brian Stoltz, and David MacMillan for their support in my career development as well as for their helpful suggestions on my research and proposals.
All the members of the Grubbs group are wonderful. I appreciate Jen Love, JP Morgan, Professor So-Yeop Han, Tae-Lim Choi, and James Tsai for the kind help when I first joined the group and embarked on a journey here. I thank Professor Han especially for introducing the Grubbs group to me and strongly recommending that I join the group. It did not take me so long to realize that she was right.
v Professor Louis Kuo, and Pawel Sledz, we have challenged aqueous metathesis and applications. I thank Jason for letting me get involved in the PEG project and helping me work on aqueous metathesis. I am grateful to Professor Louis Kuo at Lewis & Clark College for participating in my career development with good advice and collaboration in aqueous projects. I thank Pawel Sledz for working with me as a SURF student. It was good to see his improvements and build our friendship. I hope he will be a great chemist in the near future as he wishes. I thank Anatoly Chlenov for letting me get involved in the interesting CH activation project with his catalyst. The independent research from Anna Wenzel and Tina Salguero made my decomposition work look beautiful with cool, novel organometallic complexes. I deeply thank all of my collaborators for their great contributions to my research and for their helpful suggestions.
vi I am very grateful to people who have critiqued my proposals and manuscripts and proofread my English writings—Donde, Jen, Jason, Chris Douglas, Anna, Tina, Louis, Dan, Katie Campbell, Georgios, Cheol, Paula Diaconescu, Anatoly, Greg Beutner, Erin, Daryl Allen, and Mike Page. Their invaluable help and suggestions have reflected on all of my publications including this dissertation.
I appreciate Caltech staff for their kind support. I especially thank Linda Syme, a wonderful manager of the Grubbs group, Dian Buchness, Tom Dunn, Laura Howe, Steve Gould, and Rick Gerhard. I also thank Larry Henling and Dr. Mike Day for X-ray crystallography, Dr. Mona Shahgholi for mass spectroscopy, Dr. Scott Ross, Dr. Sonjong Hwang and Jon Owen for their helpful suggestions on NMR experiments.
I want to thank people at Smyrna Church and the Caltech Korean Association. I deeply appreciate Pastor Seong-Soo Kim for his sermons and help. I have never met such a pastor who clearly knows and teaches the Gospel, the meaning of life in this world for Christians, and how and for what we should live, solely based on the Bible. I appreciate all Smyrna church members who have shown true love to my family. There are too many people to whom I owe the debt of love at church. All I can say is I truly thank and love you with the love of Jesus Christ. I also appreciate Korean school teachers who voluntarily teach lovely children every Saturday morning. I also have had a great time talking about politics, science policies, sciences, and news with Caltech Korean folks. Thank you all!
vii them all, and hope to get together with them more frequently in the future with my pretty nephew and nieces.
Most of all, I want to thank my wife, Jamie (Ja-Young) and my son, David (Joon-Pyo). Meeting Jamie and David have been the most fortunate and wonderful events during my graduate study and during my whole life. I deeply thank Jamie for her bottomless support and love. I hope she will become a good dentist who truly cares about her patients. With David, my lifestyle has been totally changed. Sometimes, it was difficult to balance out family life and study, but David’s million-dollar smile was more than enough to get me going.
viii
Abstract
The recent development of ruthenium olefin metathesis catalysts, which show high activity and functional group tolerance, has expanded the scope of olefin metathesis. To improve efficiency of the ruthenium-catalyzed olefin metathesis, this dissertation describes: (1) mechanistic study to understand decomposition pathways of ruthenium olefin metathesis catalysts for the development of more stable and efficient catalysts, (2) a method to prevent an undesirable side reaction for the improvement of selectivity of ruthenium-catalyzed olefin metathesis, and (3) a novel ruthenium catalyst to increase olefin metathesis efficiency in aqueous media for potential biological applications and environmentally friendly approaches to this chemistry.
ix
Table of Contents
Chapter 1. Introduction 1
References and Notes 5
Chapter 2. Decomposition of (H2IMes)(PCy3)(Cl)2Ru=CH2, A Key Intermediate in Ruthenium Catalyzed Olefin Metathesis
Abstract 6
Introduction 6
Results and Discussion 9
Conclusion 15
Experimental 15
References and Notes 18
Chapter 3. Decomposition of Ruthenium Olefin Metathesis Catalysts
Abstract 21
Introduction 22
Results and Discussion 23
Conclusion 36
Experimental 37
References and Notes 51
Chapter 4. Double CH Activation of an N-Heterocyclic Carbene Ligand in a Ruthenium Olefin Metathesis Catalyst
Abstract 55
Introduction 55
Results and Discussion 56
Conclusion 61
x
References and Notes 65
Chapter 5. Prevention of Undesirable Olefin Isomerization during Olefin Metathesis
Abstract 67
Introduction 67
Results and Discussion 68
Conclusion 74
Experimental 75
References and Notes 83
Chapter 6. Highly Active Water-Soluble Olefin Metathesis Catalyst
Abstract 86
Introduction 86
Results and Discussion 88
Conclusion 93
Experimental 93
References and Notes 98
Chapter 7. Efficient Removal of Ruthenium Byproducts from Olefin Metathesis Products by Simple Aqueous Extraction
Abstract 101
Introduction 101
Results and Discussion 102
Conclusion 106
Experimental 106
Chapter 1
Introduction
Olefin metathesis is a powerful carbon-carbon bond formation reaction in both polymer and small molecule synthesis.1,2 This reaction is a unique process of carbon-carbon double bond rearrangement mediated by transition-metal catalysts. This reaction has an enormous variety of applications, including ring-closing metathesis (RCM), cross metathesis (CM), acyclic diene metathesis polymerization (ADMET), ring-opening metathesis polymerization (ROMP), and ring-opening cross metathesis (ROCM) (Figure 1). The generally accepted mechanism, originally proposed by Chauvin in 1971, involves the formation and subsequent cleavage of a metallacyclobutane intermediate (Scheme 1).3
X X
X
ROMP
RCM ADMET
n
n
n
+C2
H4 +C2
H
4
R1
R2
R1 R
2
X
R X
R
CM
ROCM
Intermolecular Metathesis Reactions Diene Metathesis Reactions
+
[image:11.612.101.536.473.680.2]C2H4
Scheme 1
R1
R2
R3
[M] R2
R3
R1
[M] R2
R1 R3 [M]
Olefin metathesis was discovered in the mid-1950s independently by workers at DuPont, Standard Oil of Indiana, and Phillips Petroleum as a reaction catalyzed either homo- or heterogeneously usually by multicomponent systems that consisted of early transition metal salts and alkylating agents.4 In 1966, the Triolefin Process, conversion of propene to ethylene and butanes, was commercialized, but later discontinued by other less expensive processes.4
However, there were only a few advances made in catalyst design until the isolation of the first well-defined metal carbene complexes in the late 1970s. In particular, the titanium-based “Tebbe” complex 1 provided many of the mechanism insights on olefin metathesis.5 Schrock introduced tungsten and molybdenum alkylidene catalysts that opened a new era of olefin metathesis.6-8 A variety of Mo and W catalysts 2 were developed by Schrock in the late 1980s and early 1990s. These complexes enabled living ROMP reactions and RCM applications.9-11
Ti C H2
AlCH3 CH3 Cl M t-Bu RO RO N i-Pr i-Pr
In spite of these advances, low thermal stability and oxophilicity of early transition metals limited the scope of substrates in Mo- and W-catalyzed olefin metathesis. In 1992, Grubbs and co-workers reported the first well-defined ruthenium-carbene olefin metathesis catalyst.12 Since then, a wide variety of ruthenium-based catalysts have been developed. In particular, ruthenium olefin metathesis catalysts 36 have been used extensively due to their high activity and functional group tolerance. Over the last decade, the applications of ruthenium-based olefin metathesis catalysts have expanded to include the synthesis of molecules in organic, inorganic, biochemical, polymer, and materials chemistry.13-16
Ru PCy3 Ph Cl Cl Ru PCy3 PCy3 Ph Cl Cl Ru Cl O Cl Ru N Cl Cl N Ph
N N N N N N
3 4 5 6
However, there are still many challenges in olefin metathesis that include catalyst stability, substrate specificity, stereoselectivity, enantioselectivity, use of large amount of solvent for RCM, metathesis in aqueous media, and residual metal removal.17
References and Notes
(1) Ivin, K. J.; Mol, J. C. Olefin Metathesis and Metathesis Polymerization; Academic Press: San Diego, CA, 1997.
(2) Handbook of Metathesis; Grubbs, R. H., Ed.; Wiley-VCH: Weinheim, Germany, 2003; Vol. 1-3.
(3) Herisson, J. L.; Chauvin, Y. Makromol. Chem. 1971, 141, 161.
(4) Spessard, G. O.; Miessler, G. L. Organometallic Chemistry; Prentice-Hall, Inc.: Upper Saddle River, New Jersey, 1997.
(5) Tebbe, F. N.; Parshall, G. W.; Reddy, G. S. J. Am. Chem. Soc. 1978, 100, 36113613. (6) Schrock, R. R. Angew. Chem., Int. Ed. 2006, 45, 37483759.
(7) Schrock, R. R. Acc. Chem. Res. 1990, 23, 158165.
(8) Feldman, J.; Schrock, R. R. Prog. Inorg. Chem. 1991, 39, 174. (9) Gilliom, L. R.; Grubbs, R. H. J. Am. Chem. Soc. 1986, 108, 733742.
(10) Schrock, R. R.; Feldman, J.; Cannizzo, L. F.; Grubbs, R. H. Macromolecules 1987, 20, 11691172.
(11) Grubbs, R. H.; Miller, S. J.; Fu, G. C. Acc. Chem. Res. 1995, 28, 446452.
(12) Nguyen, S. T.; Johnson, L. K.; Grubbs, R. H.; Ziller, J. W. J. Am. Chem. Soc. 1992, 114, 39743975.
(13) Trnka, T. M.; Grubbs, R. H. Acc. Chem. Res. 2001, 34, 1829. (14) Grubbs, R. H. Tetrahedron 2004, 60, 71177140.
(15) Fürstner, A. Angew. Chem. Int. Ed. 2000, 39, 30123043.
Chapter 2
Decomposition of (H
2IMes)(PCy
3)(Cl)
2Ru=CH
2, A Key Intermediate in
Ruthenium Catalyzed Olefin Metathesis
Abstract
Dinuclear ruthenium complex with a bridging carbide and a hydride ligand, and methyltricyclohexylphosphonium chloride result from thermal decomposition of olefin metathesis catalyst, (H2IMes)(PCy3)(Cl)2Ru=CH2. Involvement of dissociated phosphine in the decomposition is proposed. The dinuclear complex has catalytic olefin isomerization activity, which can be responsible for competing isomerization processes in certain olefin metathesis reactions.
Introduction
Over the past decade, olefin metathesis has emerged as a powerful method for the formation of carbon-carbon double bonds.1,2 In particular, ruthenium olefin metathesis catalysts, such as 1 and 2, have been used extensively in organic and polymer chemistry due to their high activity and functional group tolerance.3-6
Portions of this chapter have been published: Hong, S. H.; Day, M. W.; Grubbs, R. H. J.
Ru PCy3 Ph Cl N N Cl Ru PCy3 PCy3 Ph Cl Cl Ru PCy3 Cl N N Cl
1 2 3 4
CH2 Ru PCy3 PCy3 CH2 Cl Cl
Despite these advances, one of the major limiting factors for the use of ruthenium carbene catalysts in many reactions is the lifetime and efficiency of these catalysts. As a result, the ring closing of large rings requires high-dilution conditions, and the metathesis of highly substituted or electron-deficient olefins still requires elevated temperatures and extended reaction times.7-10 Furthermore, catalyst decomposition sometimes leads to unwanted side reactions, such as olefin isomerizations.11-14 As identified in previous work, the key to catalyst efficiency is the ratio of the rate of olefin metathesis relative to that of catalyst decomposition.15 Thus to rationally design a more efficient catalyst for olefin metathesis, it is essential to understand the decomposition pathways of existing catalysts.
In spite of the information obtained from kinetic studies, it has been difficult to understand the decomposition pathway of the ruthenium olefin metathesis catalysts since there has been no report of well-characterized decomposition products generated under typical metathesis conditions. Fortuitously, during the synthesis of 2, we observed a decomposition product 5.19 In complex 5, the Ru center has inserted into a CH bond of one of the mesityl groups. It has also been observed that complexes 1 and 2 decompose into hydrido-carbonyl-chloride complexes 6 and 7 upon treatment with methanol.19-22 Diver and co-workers reported carbon monoxide-promoted benzylidene or methylidene insertion into a mesityl group of complex 2 or 4.23 Van Rensburg and co-workers have suggested a substrate-induced decomposition mechanism for these catalysts based on DFT calculations involving a
-hydride transfer from a ruthenacyclobutane intermediate.24 However, the lack of well-characterized decomposition products under typical metathesis conditions has limited the understanding of the decomposition mechanism overall. In this chapter, the first well-characterized decomposition products of the N-heterocyclic based ruthenium olefin metathesis catalyst 4 in benzene is reported.
Ru
PCy3
Cl N N
CO
5
Ru
PCy3
Cl N N
CO Ru
PCy3
PCy3
Cl
CO
6 7
Results and Discussion
Thermal decomposition of complex 4 was studied. When complex 4 was monitored at 55 oC in C6D6 by 31P NMR spectroscopy, a new peak at 34.5 ppm was observed. This peak increased over time while the peak corresponding to methylidene 4 (38.6 ppm) decreased. An orange-yellow crystalline solid precipitated from the solution as decomposition proceeded, and it was isolated as 8 in 46% yield after 72 h. Formation of complex 8 is reproducible in benzene solution (Scheme 1).
Scheme 1
C6H6 55 oC
+ CH3PCy3+Cl
-8 Ru Ru C N N Mes Mes Cl Cl Cl H Ru PCy3 Cl N N Cl CH2 N N 0.023 M
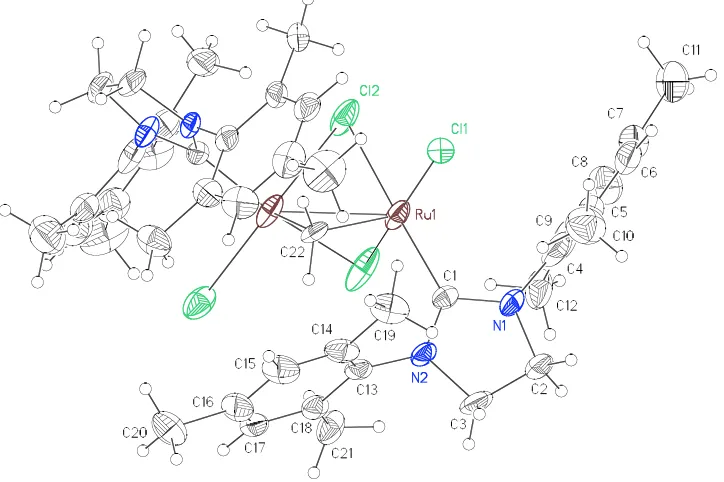
Figure 1. ORTEP drawing of 8 with thermal ellipsoids at 50% probability.
[image:20.612.119.511.411.684.2]The carbide between the ruthenium centers has a distinctive 13C chemical shift of 414.0 ppm, coupled with the hydride (JHC = 10.4 Hz); this falls within the range of 211446.3 ppm known for other μ-carbide complexes.25-28 The single carbon bridge between two ruthenium centers is slightly bent with a Ru1C22Ru2 angle of 160.3(2)o. The Ru1C22 distance in 8 is 1.698(4) Å and is slightly longer than in other reported μ-carbide ruthenium complex such as in (PCy3)2(Cl)2RuCPd(Cl)2(SMe2) (1.662(2) Å)25 and [(PPri3)2(Cl)(CF3CO2)RuCCH2Ph][BAr4] (1.660(4) Å).29 The Ru2C22 distance of 1.875(4) Å is much shorter than the usual RuC single bonds in ruthenium complexes with carbide ligands that generally range from 2.00 to 2.09 Å30-32 such as in [(Me3CO)3WCRu(CO)2(Cp)] (2.09(2) Å).32 Although the allylidene alternative [Ru=C=Ru] is possible on the basis of bond lengths, we assign the Ru1C22 interaction as a triple bond and the Ru2C22 interaction as a single bond based on the electron distribution on the ruthenium atoms. If considering the allylidene alternative, the Ru1 and Ru2 would be 15- and 19-electron species, respectively.
crystals of 9 were observed with yellow-orange crystals of 4 from a saturated benzene solution of 4 at room temperature after two weeks under an N2 atmosphere.
Scheme 2. A proposed mechanism.
Ru PCy3 Cl N N Cl CH2 Ru N N CH2 PCy3 Ru Cl N N Cl H2 C PCy3 Ru Cl N N
Cl PCy3
Ru Cl N N Cl PCy3 CH2 Ru Cl N N Cl Ru N N CH2
H2C PCy3 H3C PCy3
Cl -Ru Ru C N N Mes Mes Cl Cl Cl H N N +
+ N Ru Ru
N Mes Mes Cl Cl Cl N N CH2 Cl Ru Ru N N Mes Mes Cl Cl N N CH Cl
H2C PCy3
10 11 9 8 12 13 k1 k-1 k2 Cl2 Cl2
explained by oxidative addition of the terminal alkylidyne in 13 with migration of two chlorides. However, none of these intermediates has been observed by NMR spectroscopy.
Application of the steady-state approximation to 10 affords the decomposition rate expression (equation 1), assuming the phosphine-attacking step is rate determining. This expression is consistent with the independence of phosphine concentration and the first order kinetic observation on the decomposition of 4.17
Ru PCy3 Cl N N Cl
CH2 Cl Ru
N N
Cl
CH2 + PCy3 k1
k-1
Decomposition k2
rate of decomposition = k2[10][PCy3] =
k1k2[4][PCy3] k-1[PCy3] + k2[PCy3]
k1k2 k-1 + k2
= [4]
k1[4] k-1[PCy3] + k2[PCy3] [10] =
steady state approximation
(1)
4 10
To prevent the phosphine attack on the methylidene, hydrochloric acid was added in the benzene solution of 4. The half-life of 4 in 2.5 equiv. of HCl at 55 oC is shorter than that of 4 without HCl (4 h 35 min vs. 5 h 40 min, Chart 1). The methyltricyclohexylphosphonium salt 9 and complex 8 were not observed. Instead, unidentified phosphorous peaks around
Chart 1. Decomposition of 4 in the presence of HCl.
responsible for the undesirable isomerization reaction during difficult olefin metathesis reactions.
Scheme 3
1.5 mol % 8
CD2Cl2
40 oC, 1 d
76 %
(trans:cis = 8:1)
Conclusion
The dinuclear ruthenium complex 8, and methyltricyclohexylphosphonium chloride9 result from thermal decomposition of olefin metathesis catalyst 4 in benzene. It is proposed that dissociated phosphine is involved in the decomposition of 4. In addition, complex 8 has catalytic olefin isomerization activity, which can be responsible for competing isomerization processes in certain olefin metathesis reactions.
Experimental
General considerations. Manipulation of organometallic compounds was performed
using standard Schlenk techniques under an atmosphere of dry argon or in a nitrogen-filled
Vacuum Atmospheres drybox (O2 < 2 ppm). NMR spectra were recorded on a Varian Inova
(499.85 MHz for 1
H; 202.34 MHz for 31
P; 125.69 MHz for 13
C) or on a Varian Mercury 300
(299.817 MHz for 1
H; 121.39 MHz for 31
P; 74.45 MHz for 13
C). 31
P NMR spectra were
referenced using H3PO4 ( = 0 ppm) as an external standard. Elemental analyses were
600H spectrophotometer. GC spectra were recorded on Hewlett-Packard 5970B MSD with
5890 GC. Benzene, benzene-d6, pentane, and methylene chloride were dried by passage
through solvent purification columns. CD2Cl2 was dried by vacuum transfer from CaH2. All
solvents are degassed by standard procedure. Allylbenzene was obtained from Aldrich and
used as received. (IMesH2)(PCy3)(Cl)2Ru=CH2 was prepared according to literature
procedure.17
Decomposition of complex 4. Complex 4 (48.3 mg, 0.06 mmol) was dissolved in
benzene (2.7 mL) in a sealed tube. The reaction mixture was heated to 55 o
C. Precipitation of
orange-yellow crystalline solid was observed after 7 h. After 72 h, the precipitates were
filtered, washed with benzene and dried under vacuum to afford complex 8 (13.2 mg, 46%).
Methyltricyclohexylphosphonium chloride 9 was obtained along with some unidentified
decomposed ruthenium species by the addition of pentane (5 mL) to the filtered benzene
solution.
5.72; N, 6.00. Found: C, 55.58; H, 5.64; N, 5.64. HRMS analysis (FAB) m/z: Calcd [M+]: 936.1424, Found: 936.1434. Crystal data for 8: C41H53N4Cl3Ru2 • 1C6H6, M=1051.55, monoclinic, space group P21/c, a = 12.4536(9) Å, b = 16.1001(11) Å, c = 19.7050(8) Å, = 102.7980(10)o, V = 4843.9(6) Å3, T = 100(2) K, Z = 4, (Mo-K) = 0.828 mm-1, 42525 measured reflections, 7073 reflections with I > 2 (I), final R1 = 0.0488, wR2 = 0.0797803. CCDC reference number 233170.39
Methyltricyclohexylphosphonium chloride 9. 1
H NMR (C6D6): 2.61 (m, (CHC5H10)3-PCH3
+
,3H), 2.42 (d, 3H, CH3-PCy3 +
, JHP = 12.6Hz), 1.85-1.00 (m, 30H). 13
C{1 H}
NMR (C6D6): 30.43 (d, (CHC5H10)3-PCH3 +, J
CP = 42.6Hz), 27.11 (d, JCP = 3.1Hz), 26.47 (d, JCP = 12.6 Hz), 25.86, 1.5 (d, CH3-PCy3
+
, JCP = 47.6Hz). 31
P{1
H} NMR (C6D6): 34.5 ppm. HRMS analysis (FAB) m/z: Calcd for C19H36P [M
+
]: 295.2555, found: 295.2557.
References and Notes
(1) Ivin, K. J.; Mol, J. C. Olefin Metathesis and Metathesis Polymerization; Academic Press: San Diego, CA, 1997.
(2) Handbook of Metathesis; Grubbs, R. H., Ed.; Wiley-VCH: Weinheim, Germany, 2003; Vol. 1-3.
(3) Trnka, T. M.; Grubbs, R. H. Acc. Chem. Res. 2001, 34, 18-29. (4) Grubbs, R. H. Tetrahedron 2004, 60, 7117-7140.
(5) Fürstner, A. Angew. Chem. Int. Ed. 2000, 39, 3012-3043.
(6) Frenzel, U.; Nuyken, O. J. Polym. Sci., Part A: Polym. Chem. 2002, 40, 2895-2916. (7) Han, S.-Y.; Chang, S. In Handbook of Metathesis; Grubbs, R. H., Ed.; Wiley-VCH:
Weinheim, 2003; Vol. 2, p 5-127.
(8) Nicola, T.; Brenner, M.; Donsbach, K.; Kreye, P. Org. Process Res. Dev. 2005, 9, 513-515.
(9) Vinokurov, N.; Michrowska, A.; Szmigielska, A.; Drzazga, Z.; Wojciuk, G.; Demchuk, O. M.; Grela, K.; Pietrusiewicz, K. M.; Butenschon, H. Adv. Synth. Catal. 2006, 348, 931-938.
(10) Martin, W. H. C.; Blechert, S. Curr. Top. Med. Chem. 2005, 5, 1521-1540. (11) Alcaide, B.; Almendros, P. Chem. Eur. J. 2003, 9, 1259-1262.
(12) Schmidt, B. Eur. J. Org. Chem. 2004, 1865-1880.
(13) Hong, S. H.; Sanders, D. P.; Lee, C. W.; Grubbs, R. H. J. Am. Chem. Soc. 2005, 127, 17160-17161.
(15) Ulman, M.; Grubbs, R. H. J. Org. Chem. 1999, 64, 7202-7207.
(16) Hong, S. H.; Day, M. W.; Grubbs, R. H. J. Am. Chem. Soc. 2004, 126, 7414-7415. (17) Sanford, M. S.; Love, J. A.; Grubbs, R. H. J. Am. Chem. Soc. 2001, 123, 6543-6554. (18) Ulman, M. PhD Thesis, California Institute of Technology, 2000.
(19) Trnka, T. M.; Morgan, J. P.; Sanford, M. S.; Wilhelm, T. E.; Scholl, M.; Choi, T. L.; Ding, S.; Day, M. W.; Grubbs, R. H. J. Am. Chem. Soc. 2003, 125, 2546-2558.
(20) Banti, D.; Mol, J. C. J. Organomet. Chem. 2004, 689, 3113-3116. (21) Dinger, M. B.; Mol, J. C. Eur. J. Inorg. Chem. 2003, 2827-2833. (22) Dinger, M. B.; Mol, J. C. Organometallics 2003, 22, 1089-1095.
(23) Galan, B. R.; Gembicky, M.; Dominiak, P. M.; Keister, J. B.; Diver, S. T. J. Am. Chem. Soc. 2005, 127, 15702-15703.
(24) van Rensburg, W. J.; Steynberg, P. J.; Meyer, W. H.; Kirk, M. M.; Forman, G. S. J. Am. Chem. Soc. 2004, 126, 14332-14333.
(25) Hejl, A.; Trnka, T. M.; Day, M. W.; Grubbs, R. H. Chem. Commun. 2002, 2524-2525.
(26) Etienne, M.; White, P. S.; Templeton, J. L. J. Am. Chem. Soc. 1991, 113, 2324-2325. (27) Miller, R. L.; Wolczanski, P. T.; Rheingold, A. L. J. Am. Chem. Soc. 1993, 115,
10422-10423.
(28) Beck, W.; Knauer, W.; Robl, C. Angew. Chem., Int. Ed. Engl. 1990, 29, 318-320. (29) González-Herrero, P.; Weberndörfer, B.; Ilg, K.; Wolf, J.; Werner, H. Angew. Chem.,
Int. Ed. 2000, 39, 3266.
(31) Ren, T.; Zou, G.; Alvarez, J. C. Chem. Commun. 2000, 1197-1198. (32) Latesky, S. L.; Selegue, J. P. J. Am. Chem. Soc. 1987, 109, 4731-4733. (33) Bestmann, H. J.; Kratzer, O. Chem. Ber. Recl. 1962, 95, 1894.
(34) Hansen, S. M.; Rominger, F.; Metz, M.; Hofmann, P. Chem. Eur. J. 1999, 5, 557-566. (35) Tan, K. L.; Vasudevan, A.; Bergman, R. G.; Ellman, J. A.; Souers, A. J. Org. Lett.
2003, 5, 2131-2134.
(36) Lehman, S. E.; Schwendeman, J. E.; O'Donnell, P. M.; Wagener, K. B. Inorg. Chim. Acta 2003, 345, 190-198.
(37) Schmidt, B. J. Org. Chem. 2004, 69, 7672-7687.
(38) Sutton, A. E.; Seigal, B. A.; Finnegan, D. F.; Snapper, M. L. J. Am. Chem. Soc. 2002, 124, 13390-13391.
Chapter 3
Decomposition of Ruthenium Olefin Metathesis Catalysts
Abstract
The decomposition of a series of ruthenium metathesis catalysts has been examined
using methylidene species as model complexes. All of the phosphine-containing methylidene
complexes decomposed to generate methylphosphonium salts, and their decomposition
routes followed first order kinetics. The formation of these salts in high conversion, coupled
with the observed kinetic behavior for this reaction, suggest that the major decomposition
pathway involves nucleophilic attack of a dissociated phosphine on the methylidene carbon.
This mechanism also is consistent with decomposition observed in the presence of ethylene
as a model olefin substrate. The decomposition of phosphine-free catalyst
(H2IMes)(Cl)2Ru=CH(2-C6H4-O-i-Pr) (H2IMes =
1,3-dimesityl-4,5-dihydroimidazol-2-ylidene) with ethylene was found to generate unidentified ruthenium hydride species. The
novel ruthenium complex (H2IMes)(pyridine)3(Cl)2Ru, which was generated during the
synthetic attempts to prepare the highly unstable pyridine-based methylidene complex
Introduction
The recent development of ruthenium olefin metathesis catalysts such as 14, which show high activity and functional group tolerance, has expanded the scope of olefin metathesis.1-4
Ru
PCy3 Ph Cl
Cl Ru
PCy3
PCy3 Ph Cl
Cl
1 2 3 4
Ru Cl
O Cl
Ru
N Cl
Cl N
Ph
N N N N N N
Scheme 1. Decomposition of the methylidene complex 5.
C6H6 55 oC
+ CH
3PCy3+Cl -Ru Ru C N N Mes Mes Cl Cl Cl H Ru PCy3 Cl N N Cl CH2 N N
5 6 7
Scheme 2. Proposed decomposition mechanism.
Ru PCy3
Cl Cl
CH2
Ru CH2
PCy3 Ru Cl Cl H2 C PCy3 Ru Cl
Cl PCy3
Ru Cl Cl PCy3 CH2 Ru Cl Cl H
2C PCy3 H3C PCy3
Cl -Ru Ru C N N Mes Mes Cl Cl Cl H N N +
+ N Ru Ru
N Mes Mes Cl Cl Cl N N CH2 Cl Ru Ru N N Mes Mes Cl Cl N N CH Cl
H2C PCy3
8 9 10 11 k1 k-1 k2 Cl2 12 6 7
N N N N
N N N N N N
N N
Ru CH2
Cl2 N N
Results and Discussion
methylidene resonance integral over time.6 Recrystallization and spectroscopic methods were used to identify and characterize decomposition products. All of the tested methylidene complexes of phosphine-based ruthenium catalysts decomposed to generate methyltricylohexylphosphonium salts as the major phosphine species (Table 1). In our previous report, we were not able to conclusively identify the phosphine product from the decomposition of complex 13. In this case, the phosphine activation was proposed based on the 2H-NMR study with (PCy3)2Cl2Ru=CD2 (13-d2).6 Here, the product was characterized successfully as CH3PCy3+Cl- (7) by comparison with an independently prepared sample of the salt (1H, 13C, and HRMS). The 2H peak observed at ~2.5 ppm during the decomposition of 13-d2 originates from the methyl protons of CD3PCy3+Cl- (7-d3).
The conversions to the phosphine products CH3PCy3+X- were determined by comparing the 1H NMR integration of the -proton in the cyclohexyl rings of the phosphonium salts with an internal standard (anthracene). The conversions were high, 81%
Table 1. Decomposition of ruthenium methylidene complexesa
Entry Methylidenes Half-Life kdecomp
(s-1)
Decomposition Products (Conversion)b 1 Ru PCy3 PCy3 CH2 Cl Cl 13
40 min 0.016 CH3PCy3+Cl- (82%)
2 Ru PCy3 PCy3 CH2 Br Br 14
35 min 0.018 CH3PCy3 +
Br- (15, 85%)
3 Ru PCy3 CH2 Cl Cl N N 5
5 h 40 min 0.0021 6 (46%) c CH3PCy3+Cl- d
4 Ru PCy3 CH2 Br Br N N 16
5 h 15 min 0.0024 CH3PCy3+Br- d
5 Ru Cl N N Cl CH2 PCy3 17
1 h 0.011 CH3PCy3 +
Cl- (81%) H2IPrH+Cl- e
a
Conditions: 0.023M, C6D6, 55 o
C, anthracene as an internal standard. b
Determined by 1
H NMR spectroscopy. c
Isolated yield. d
Conversions could not be determined. e
H2IPr =
The decompositions of phosphine-based ruthenium methylidene complexes were found to follow first-order kinetics; the decomposition rates were not affected by the addition of excess phosphine.5-7 As anticipated, catalysts containing an N-heterocyclic carbene ligand had increased lifetimes compared with bis(phosphine)-based catalysts.5,8,9 Changing the chloride ligands to bromides was found to only slightly decrease the catalyst lifetimes. Attempts to replace the chloride ligands with iodides were unsuccessful, presumably due to even more rapid decomposition.7
Scheme 3. Phosphine dissociation and attack mechanism.
Ru L
PCy3
X
X
CH2 Ru
L
CH2 PCy3 Ru
L H2 C PCy3 Ru L PCy3 Ru L PCy3 CH2 +
H2C PCy3
k1 k-1
k2
X2 X2
X2
X2 Ru
L
X2 Decomposition
H2C PCy3 CH3PCy3+X
-[Ru] [Ru*]
rate of decomposition = k2[Ru*][PCy3] =
k1k2[Ru][PCy3] k-1[PCy3] + k2[PCy3]
k1k2 k-1 + k2
= [Ru] (1)
accelerates phosphine attack on the methylidene carbon.12 This result indicates that a mechanism involving phosphine dissociation and attack (Scheme 3) is more reasonable than a mechanism involving the internal attack of phosphine (Scheme 4).
Scheme 4. Internal phosphine attack mechanism.
Ru L
PCy3 X
X
CH2 Ru
L H2 C PCy3 Ru L P Cy3 Ru L PCy3 CH2
H2C PCy3 kdecomp
X2
X2 X2
Ru L X2
Decomposition
H2C PCy3 CH3PCy3+X -[Ru]
rate of decomposition = kdecomp[Ru] (2)
ligands.15 Because it is not, we favor the mechanism involving the nucleophilic attack of free phosphine for the decomposition of ruthenium methylidene complexes (Scheme 3). The nucleophilic attack of phosphines on the carbene carbon of ruthenium alkylidenes has also been reported by Hofmann and co-workers.16
Figure 1.31
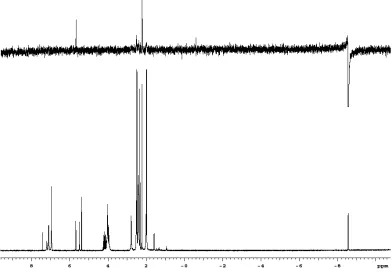
P NMR spectrum of the decomposition of 13 in the presence of ethylene.
able to characterize the major phosphine decomposition product. We have reexamined this reaction and found that 31P NMR spectra of decomposed samples reveal a major phosphine complex at 34.6 ppm after decomposition of both 5 and 13 (Figure 1), which corresponds to CH3PCy3+Cl-. The identity of this species was confirmed by spectroscopic methods. The 13C NMR spectra which shows a characteristic doublet for the phosphonium salt’s methyl protons at 1.5 ppm, was particularly revealing.5 From this evidence, we believe that phosphine attack on the methylidene carbon is also a major pathway in the decomposition of 5 and 13 in the presence of ethylene.
be in effect. However, complex 6 was not observed in the reaction mixture, suggesting a divergent mechanistic pathway. Mass spectrometric analysis (FAB+) of the reaction mixture at 8 minutes identified a ruthenium species with a m/z of 971.1, supporting the identity of the intermediate alkylidene to be complex 11, a species that had originally been proposed in the decomposition of 5 but not observed (Scheme 2).5 This intermediate appears to be capable of reverting back to a 14-electron methylidene species, based on the observation that decreasing the temperature of a reaction mixture containing 11 to 40 °Cin the presence of ethylene was found to generate metallaycle 19.21-23 It is important to note that catalyst 18 does not react with ethylene at this temperature.
Scheme 5. Reaction of catalyst 18 with ethylene.
ethylene (~1 atm) CD2Cl2 (0.035 M) Cl Ru Cl N N Ph PPh3 Ru Cl Cl N N 18
33% yield (83% conv. of 18) 1H NMR: = 18.59 ppm MS(FAB+): m/z = 971.1 (M-H) *correlates with the calculated value for 11
40 °C 3 h
19 23 °C, 8 min
Scheme 6. Isolation of the major decomposition product in the reaction of 18 with ethylene.
ethylene (~1 atm) toluene (0.035 M)
23 °C, 5 d Cl Ru Cl N N Ph PPh3 Ru N N 18 Ru N N Cl Cl 21 ~70% yield +
H3C PPh3 Cl
Scheme 7. Proposed mechanistic pathway for the decomposition of the methylidene of 18 with ethylene. Cl Ru Cl N N CH2 PPh3 Ru N N Ru N N Cl Cl 21 H3C PPh3Cl
- PPh3
+ PPh3 Cl
Ru Cl N N
CH2 + PPh3
8 Cl Ru Cl N N PPh3 Cl Ru Cl N N PPh3 Cl Ru Cl N N CH2 N N
Cl2Ru +
CH2
Ph3P x 2
23 20 Cl Ru N N CH2 Cl Ru N N 11 CH2 Ph3P
23 22
8
The major decomposition product of 18 with ethylene ultimately was identified by running the reaction in Scheme 5 on a 77 μmol scale in toluene. After 5 days at 23 °C, methyltriphenylphosphonium chloride was isolated in quantitative yield, in addition to 52 mg of a red-brown, crystalline solid that was found to be unstable in solution in the absence of ethylene. X-ray analysis determined the crystal structure to be that of the C2-symmetric complex 21 (Scheme 6), which was presumably derived from the ortho-methyl C-H activation of two ruthenium-coordinated NHC ligandsin the presence of two equivalents of methylenetriphenylphosphine (23) as a base.
appears to be more vulnerable to phosphine attack and subsequent decomposition relative to 5, resulting in the minimal population (<2%) of 22 that had been observed during the course of the reaction. Differences in this decomposition route compared to that proposed for catalyst 5 potentially can be attributed to the weaker basicity and less steric hindrance of triphenylphosphine relative to tricyclohexylphosphine24,25 as well as the presence of ethylene in the reaction mixture.
[image:42.612.136.498.435.676.2]Other decomposition products observed during synthesis of complex 5. During the synthesis of methylidene 5 in the atmosphere of 11.5 atm of ethylene gas, the ruthenium dimer complex 24 was observed (Scheme 8). As shown by X-ray crystallography (Figure 2), this complex has a Ru—Ru single bond (2.7021(5) Å) and two bridging chlorides along with bridging methylene.26 Formation of complex 24 can be also explained as the dimerization between complex 8 and 10 generated by phosphine-involved decomposition.
Scheme 8
CH2=CH2
Benzene 50 oC, 90 min
Ru Cl Cl C H2 Ru Cl
Cl H2IMes MesIH2
+
3540% trace (<~5%) Ru PCy3 Ph Cl Cl N N Ru PCy3 CH2 Cl Cl N N 24 Scheme 9
CD2=CD2 ?
[image:43.612.88.536.84.671.2]Benzene 50 oC, 90 min Ru PCy3 Ph Cl Cl N N Ru PCy3 Cl Cl N N CO 25
Another decomposition product 25 was characterized by X-ray crystallography during an attempt to synthesize deuteriated methylidene using CD2=CD2 and catalyst 2 (Scheme 9 and Figure 3). However, the formation of 25 was not reproducible in other attempts.The purity of the used ethylene-d4 gas is suspected.27
Decomposition of phosphine-free catalysts. Catalyst 3 is known as a more stable catalyst than 2 under air and water due to the chelation of its isopropoxy ligand.28,29 However, as with the phosphine-containing catalysts, a comparison of stability between initiators is not particularly meaningful.5,6 Both catalyst 2 and 3 are thermally stable—their half-lives at 55 oC in benzene are over a month. Because the methylidene derivative of 3 cannot be isolated, its decomposition was examined directly in the presence of ethylene (Scheme 10). After one day, unidentified ruthenium hydride species were observed by 1H NMR at -1.54 and -4.96 ppm. Attempts to isolate these species were unsuccessful. These species could be responsible for the olefin-isomerization reactions known to be catalyzed by 3.30,31 This result suggests that other decomposition modes, which are only slightly slower than the phosphine-involved decomposition, are also available when a phosphine is not present.
Scheme 10. Catalyst 3 with ethylene.
Ru Cl
N N
O Cl
H2C CH2
Ru N N
CH2 Cl2
Decomposition
3
Hydrides observed (1H NMR :-1.54, -4.96 ppm)
1 atm, 55 oC
C6D6
Bispyridine-based catalysts, such as 4, have proven to be useful for the synthesis of polymers due to their fast-initiation rates.18,32 However, the lower stabilities of these catalysts limit their application. We tried to synthesize (H2IMes)(py)2(Cl)2Ru=CH2 (26) to compare with other methylidene complexes; however, any synthetic attempts were unsuccessful due to its instability. Even in situ, the methylidene protons of complex 26 were never observed by 1
H NMR.
Interestingly, complex 27 and methyltricyclohexylphosphonium chloride were formed from the reaction of 5 with an excess of pyridine (Scheme 11).33 The structure of 27 was determined by X-ray crystallography (Figure 4). All bond distances and angles in this structure are typical, but the mesityl groups are twisted by ~25o with respect to each other, which contrasts with their usual orientation perpendicular to the imidazolidine ring.
Scheme 11. Formation of (H2IMes)(py)3(Cl)2Ru (27).
Ru
PCy3 Cl
N N
Cl CH2
N excess pyridine
Ru
N Cl
N N
Cl N
27
+ CH3PCy3+Cl
[image:46.612.85.540.111.499.2]-5
Figure 4. Crystal structure of (H2IMes)(py)3(Cl)2Ru (27).
Conclusion
to generate methylphosphonium salts. The observed kinetic behavior suggests that the major decomposition pathway involves attack of the dissociated phosphine on the methylidene carbon. Such a mechanism also explains the decomposition observed in the presence of ethylene as a model olefin substrate. The novel ruthenium ethylene complex 21 was observed from the decomposition of the catalyst 18 under ethylene. The decomposition of phosphine-free catalyst 3 generated unidentified ruthenium hydride species under an atmosphere of ethylene. Attempts to synthesize a pyridine-coordinated analog of methylidene 4 were unsuccessful presumably due to rapid decomposition. Instead, we observed the tris(pyridine) complex 27 as a decomposition product. This decomposition study will provide rational basis to design and synthesize more efficient ruthenium olefin metathesis catalysts.
Experimental
degassed by either a generous Ar sparge or three freeze-pump-thaw cycles. Catalysts 1, 2, and 3 were obtained from Materia and used as received. Ruthenium complexes 4,185,713,37 18,7 and (H2IMes)(PCy3)(Br)2Ru=CHPh7 were prepared according to literature procedures. Methyltriphenylphosphonium chloride was purchased from Aldrich.
(PCy3)2(Br)2Ru=CH2 (9). A solution of 1 (166 mg, 0.182 mmol) in CH2Cl2 (3 mL) was stirred under an atmosphere of ethylene for 30 min at room temperature. The solvent was removed under vacuum, and the residue was repeatedly washed with cold pentane (5 mL) and dried under vacuum. A burgundy microcrystalline solid (146 mg, 0.175 mmol, 96%) was obtained. 1H NMR (C6D6): 19.38 (s, 2H), 3.00-2.80 (m, 6H), 1.95-1.20 (all m, 60H). 13C{1H} NMR (CD2Cl2): 297.3 (t, JCP = 8.2 Hz), 31.6 (t, JCP = 10.1Hz), 29.7 (s), 28.0 (t, JCP = 5.1 Hz), 26.8(s). 31P{1H} NMR (C6D6): 44.51 (s). HRMS analysis (FAB) m/z: Calcd for C37H68Br2P2Ru [M+]: 836.2199, found: 836.2174.
21.0 (d, JCP = 2.6 Hz), 20.9, 19.7. 31P{1H} NMR (C6D6): 38.02 (s). Anal. Calcd for C40H61N2Br2PRu: C, 55.75; H, 7.13; N, 3.25. Found: C, 56.04; H, 7.13; N, 3.25.
(H2IPr)(PCy3)(Cl)2Ru=CH2 (11). A solution of (H2IPr)(PCy3)(Cl)2Ru=CHPh (300 mg, 0.321 mmol) in C6D6 (5 mL) was stirred under an atomosphere of ethylene for 30 min at 45 oC. The brown solution was cooled to room temperature, and the product was purified by column chromatography (gradient elution: 100% pentane to 12:1 pentane/diethyl ether) to afford an orange-yellow solid (95 mg, 0.110 mmol, 34%). This product was very air-sensitive, and even slowly decomposed in the dry box. Further study was done immediately after the synthesis. 1H NMR (C6D6): 18.22 (s, 2H), 7.22-7.09 (m, 11H), 4.10 (m, 2H), 3.74-3.60 (m, 6H), 2.37-2.20 (m, 3H), 1.70-0.96 (m, 54H). 13C{1H} NMR (C6D6): 294.8 (d, JCP = 8.3 Hz), 224.2 (d, JCP = 75.7Hz), 150.1, 148.7, 137.9, 136.2, 130.9, 130.0, 128.9, 128.7, 128.5, 128.3, 125.1, 124.9, 55.2, 53.6, 31.2, 31.0, 29.6, 29.5, 28.8, 28.3, 28.3, 27.6, 27.0, 25.2, 24.3 31P{1H} NMR (C6D6): 38.83 (s). HRMS analysis (FAB) m/z: Calcd for C46H73N2Cl2RuP [M+]: 856.3932, found: 856.3917.
Complex 24. Crystal data for 24: C43H54N4Cl4Ru2 • C6H6, M=1009.90, tetragonal, space group P-421c, a = 15.4853(5) Å, c = 19.7050(8) Å, V=4725.2(3) Å3, T = 100(2) K, Z = 4, (Mo-K) = 0.900 mm-1, 76914 measured reflections, 8346 unique, 5377 reflections with I > 2 (I), final R1 = 0.0515, wR2 = 0.0797. CCDC reference number 231270.
V=4378.5(2) Å3, T = 100(2) K, Z = 4, (Mo-K) = 0.552 mm-1, 80614 measured reflections, 21479 unique, 13014 reflections with I > 2 (I), final R1 = 0.0463, wR2 = 0.0692. CCDC reference number 231269. 1H NMR (C6D6): 6.69 (s, 4H), 3.22 (m, 4H), 2.61 (s, 6H), 2.55 (s, 6H), 2.50 (m, 3H), 2.05 (s, 3H), 2.02 (s, 3H), 1.99 – 1.00 (m, 30H). 31P{1H} NMR (C6D6): 32.91.
(H2IMes)(py)3(Cl)2Ru (27). A solution of 5 (150 mg, 0.195 mmol), excess pyridine (0.25 mL), and 1.0 mL of toluene was stirred at room temperature for 30 min. 20 mL of hexanes were added, and the solution was allowed to sit without stirring for 3 min. The red solution was decanted away from the pale yellow precipitate and cooled to 0 oC. The resulting red precipitate was collected and redissolved in a minimum amount of toluene. Again, 20 mL of hexanes were added, the solution cooled, and the red precipitate collected. This procedure was repeated three more times. Finally, the precipitate was dried under vacuum to provide 0.041 g (mmol, 29%) of 27 as a red-orange solid. 1H NMR (CD2Cl2): 9.00 (d, J = 5.5 Hz, 4H), 8.70 (d, J = 5.5, 2H), 7.30 (t, J = 7.5 Hz, 1H), 6.98 (t, J = 7.5 Hz, 2H), 6.76 (t, J = 7.0 Hz, 2H), 6.34 (s, 4H), 6.33 (t, J = 7.0, 4H), 3.98 (s, 4H), 2.47 (s, 12H), 2.02 (s, 6H). 13C{1H} NMR (C6D6): 198.2, 142.2, 138.5, 128.4, 125.6, 125.0, 123.1, 120.3, 118.4, 113.1, 118.9, 54.1, 25.7, 24.5. HRMS analysis (FAB) m/z: Calcd for C36H41N5Cl2Ru [M+]: 715.1783, found: 715.2783.
used to seal the NMR tube, and this seal was reinforced with parafilm. The sample was placed into the spectrometer and allowed to equibrate at the probe temperature for 10 min. Complex decomposition was following by monitoring the diminution of the methylidene protons through collection of a time-delayed array of 1H NMR spectra (referred to as a preacquisition delay, PAD, by Varian software). Plots of [methyliene] vs. time and 31P spectra of the decompositions are shown in Charts 14 and Figures 59.
Chart 1. Decomposition of 5.
Chart 2. Decomposition of 14.
Chart 4. Decomposition of 17.
Figure 5. 31
Figure 6. 31
[image:54.612.104.524.388.666.2]P NMR spectrum after decomposition of 13.
Figure 7. 31
Figure 8. 31
P NMR spectrum after decomposition of 16.
Figure 9. 31
[image:55.612.104.520.383.681.2]Chart 5. Decomposition reaction of 18 with ethylene.
HRMS confirmed the presence of methyltriphenylphosphonium chloride (20) via correlation to authentic material.
Figure 6. Mass spectrum of the reaction of 18 with ethylene after 8 min at 23 °C.
Reaction of 18 with ethylene to generate metallacyclobutane 19.34 To a J-Young tube in the glove box, complex 18 (17.5 mg, 0.021 mmol, 1.0 equiv.) was dissolved in 600
the predominant alkylidene species (~44% yield relative to Ru). The reaction was then cooled to 40 °C. After 3 h at - 40 °C, the peak at 18.59 ppm (corresponding to complex 11), had completely diminished and two new peaks at 6.64 ppm (4H) and 2.63(2H) were clearly visible, corresponding to the literature values for the - and -hydrogens of ruthenium metallacycle 19.21,22
Table 2. Summary of crystallographic data for 7, 21 and 27
7 21 27
formula [C19H36P] +
Cl
3(H2O) C46H58N4Cl2Ru2 C36H41N5Cl2Ru
Mr 384.95 940.00 715.71
crystal color colorless red/brown orange
crystal size (mm) 0.30 0.23 0.18 0.21 0.18 0.07 0.33 0.28 0.08
crystal system triclinic triclinic orthorhombic
space group P-1 P-1 Pbcn
a (Å) 9.8774(4) 9.8735(6) 11.3376(7)
b (Å) 10.0035(5) 10.7053(7) 13.3755(8)
c (Å) 12.7700(6) 11.8315(7) 21.4203(14)
(deg) 85.1580(10) 100.828(2) 90
(deg) 74.3040(10) 98.018(2) 90
(deg) 63.5350(10) 116.4470(10) 90
V (Å3) 1086.48(9) 1063.77(11) 3248.3(4)
Z 2 1 4
Dcalcd (g cm-3) 1.177 1.467 1.463
T (K) 100(2) 100(2) 98(2)
(Å) 0.71073 0.71073 0.71073
(mm-1) 0.263 0.872 0.681
R1a(all data) 0.0618 0.0724 0.0481
wR2b (all data) 0.0772 0.0714 0.0509
GOF 1.307 1.146 1.716
a
References and Notes
(1) Handbook of Metathesis; Grubbs, R. H., Ed.; Wiley-VCH: Weinheim, Germany, 2003; Vol. 1-3.
(2) Connon, S. J.; Blechert, S. Angew. Chem., Int. Ed. 2003, 42, 1900-1923. (3) Trnka, T. M.; Grubbs, R. H. Acc. Chem. Res. 2001, 34, 18-29.
(4) Grubbs, R. H.; Chang, S. Tetrahedron 1998, 54, 4413-4450.
(5) Hong, S. H.; Day, M. W.; Grubbs, R. H. J. Am. Chem. Soc. 2004, 126, 7414-7415. (6) Ulman, M.; Grubbs, R. H. J. Org. Chem. 1999, 64, 7202-7207.
(7) Sanford, M. S.; Love, J. A.; Grubbs, R. H. J. Am. Chem. Soc. 2001, 123, 6543-6554. (8) Ulman, M. PhD Thesis, California Institute of Technology, 2000.
(9) Huang, J. K.; Schanz, H. J.; Stevens, E. D.; Nolan, S. P. Organometallics 1999, 18, 5375-5380.
(10) Fürstner, A.; Ackermann, L.; Gabor, B.; Goddard, R.; Lehmann, C. W.; Mynott, R.; Stelzer, F.; Thiel, O. R. Chem. Eur. J. 2001, 7, 3236-3253.
(11) Dinger, M. B.; Mol, J. C. Adv. Synth. Catal. 2002, 344, 671-677.
(12) Courchay, F. C.; Sworen, J. C.; Wagener, K. B. Macromolecules 2003, 36, 8231-8239.
(13) Steady-state appromimation was applied to equation (1).
(14) Several unidentified phosphorus peaks were observed in 31P NMR spectra from the reaction between 5 and PMe3.
52 Scott, N. M.; Costabile, C.; Cavallo, L.; Hoff, C. D.; Nolan, S. P. J. Am. Chem. Soc.
2005, 127, 2485-2495.
(16) Hansen, S. M.; Rominger, F.; Metz, M.; Hofmann, P. Chem. Eur. J.1999, 5, 557-566.
(17) van Rensburg, W. J.; Steynberg, P. J.; Meyer, W. H.; Kirk, M. M.; Forman, G. S. J.
Am. Chem. Soc.2004, 126, 14332-14333.
(18) Sanford, M. S.; Love, J. A.; Grubbs, R. H. Organometallics2001, 20, 5314-5318.
(19) This part of decomposition study of catalyst 18 was done and written by Dr. Anna G.
Wenzel.
(20) Values correspond to NMR yields utilizing anthracene (0.014 M) as an internal
reaction standard. Alkylidene assumed to be methylidene-derived (2H); conversion
based on Ru is 66% if it is assumed that the complex is the bis-ruthenium complex
11.
(21) Romero, P. E.; Piers, W. E. J. Am. Chem. Soc.2005, 127, 5032-5033.
(22) Wenzel, A. G.; Grubbs, R. H. J. Am. Chem. Soc.2006, 128, 16048-16049.
(23) Romero, P. E.; Piers, W. E. J. Am. Chem. Soc.2007, 129, 1698-1704.
(24) Zhang, X. M.; Bordwell, F. G. J. Am. Chem. Soc.1994, 116, 968-972.
(25) Streitwieser, A.; McKeown, A. E.; Hasanayn, F.; Davis, N. R. Org. Lett. 2005, 7,
1259-1262.
(26) For diruthenium μ-methylene complexes see : Gao, Y.; Jennings, M. C.; Puddephatt,
R. J. Organometallics2001, 20, 1882.
(27) The gas used at the synthesis was purchased at Aldrich. In another trial using other
(28) Garber, S. B.; Kingsbury, J. S.; Gray, B. L.; Hoveyda, A. H. J. Am. Chem. Soc. 2000, 122, 8168-8179.
(29) Hong, S. H.; Grubbs, R. H. J. Am. Chem. Soc. 2006, 128, 3508-3509.
(30) Hong, S. H.; Sanders, D. P.; Lee, C. W.; Grubbs, R. H. J. Am. Chem. Soc. 2005, 127, 17160-17161.
(31) Courchay, F. C.; Sworen, J. C.; Ghiviriga, I.; Abboud, K. A.; Wagener, K. B. Organometallics 2006, 25, 6074-6086.
(32) Love, J. A.; Morgan, J. P.; Trnka, T. M.; Grubbs, R. H. Angew. Chem., Int. Ed. 2002, 41, 4035-4037.
(33) The reaction in Scheme 9 and characterization of complex 25 were carried out by Dr. Tina T. Salguero.
(34) Hejl, A.; Grubbs, R. H. Unpublished results.
(35) Williams, J. E.; Harner, M. J.; Sponsler, M. B. Organometallics 2005, 24, 2013-2015. (36) Methylpyridium salt was not observed in the reaction of 4 in the presence of ethylene.
Werner and co-workers have reported that a similar reaction between the ruthenium methylidene complex (PPri2Ph)2(CO)(Cl)(H)Ru=CH2 and pyridine yields (PPri2Ph)2(py)(CO)(Cl)(H)Ru. See: Werner, H.; Stuer, W.; Weberndorfer, B.; Wolf, J. Eur. J. Inorg. Chem. 1999, 1707-1713.
(37) Schwab, P.; Grubbs, R. H.; Ziller, J. W. J. Am. Chem. Soc. 1996, 118, 100-110.
(39) The presence of 11 has also been observed via mass spectrometry in the reaction of the bispyridyl catalyst 4 with ethylene.
Chapter 4
Double C
H Activation of an N-Heterocyclic Carbene Ligand in a
Ruthenium Olefin Metathesis Catalyst
Abstract
Decomposition of (BIPh)(PCy3)(Cl)2Ru=(H)Ph (BIPh = N,N’
-diphenylbenzimidazol-2-ylidene) results in the benzylidene insertion into an ortho CH bond of a BIPh N-phenyl
group. Ruthenium further inserts into another ortho CH of the other BIPh N-phenyl ring to
give a new RuC bond as a part of a five-membered metallacycle.
Introduction
N-heterocyclic carbene (NHC) ligands have been widely used for transition metal
catalysts in a role analogous to that of phosphines and other neutral two-electron donors due
to their distinctive high -basicity and low -acidity.1-3 However, it has been demonstrated
that NHCs occasionally participate in unanticipated side reactions, such as CC and CH
activation4,5
and sometimes enter into abnormal binding modes.6,7
Because these reactions
can be detrimental to catalyst function, understanding them is of fundamental importance to
the design of stable transition-metal catalysts with NHC ligands.8
In this chapter, novel
Results and Discussion
NHC-based olefin metathesis catalyst 4 shows high activity in ring-closing metathesis (RCM) reactions to form tetrasubstituted olefins.9 Although catalyst 3, a phosphine analog of 4, is also active in the RCM reactions,10 it has been found to decompose much faster having a shorter half-life than both catalysts 1 and 2 (30 min for 3 vs. 8 d for 1 and ~38 d for 2 at 55 oC in 0.023M C6D6 solution).11,12 Although it is well documented that NHC-based ruthenium olefin metathesis catalysts are generally more stable than bisphosphine-based catalysts,13 complex 3 is even much less stable than 1. This abnormal instability of 3 led us to study the structural feature of the N,N’-diphenylbenzimidazol-2-ylidene (BIPh) ligand that lead to the decomposition of 3.
Ru
PCy3 Ph Cl
Cl
1 2 3 4 Ru Cl
O Cl
N N N N
Ru PCy3 Ph Cl Cl N N Ru PCy3 PCy3 Ph Cl Cl
When we investigated the thermal stability of complex 3 in benzene solutions at 60 o
C under inert conditions, the decomposition product 5 precipitated with an isolated yield of 58% after 3 days (Scheme 1). Complex 6 was also observed in traces (<2%). Interestingly, complex 6 was the major decomposition product along with 5 after 12 hours in CD2Cl2 at 40 o
Scheme 1. Thermal decomposition of complex 3.
C6H6 60 oC, 3 d
58 % trace
CD2Cl2 40 oC, 12 h
24 % 38 % 5 6 Ru PCy3 Ph Cl Cl N N Ru PCy3 Ph Cl Cl N N Ru Cl Cl N N Ru Cl N N Ru Cl Cl N N Ru Cl N N
Figure 1. ORTEP drawing of 5. Atoms are represented by ellipsoids at the 50% probability level. Hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (deg): RuC(1), 2.042(2); RuCl(1), 2.4108(5); RuPhCenter, 1.69; C(1)RuPhCenter, 127.8; Cl(1)Ru(1)PhCenter, 126.0; C(1)RuCl(1), 86.55(5); Cl(1)RuCl(2), 85.280(18).
other ruthenium complexes.4,16,17 The planes of phenyl rings of BIPh become approximately perpendicular to allow the formation of the five-membered metallacycle.
Figure 2. ORTEP drawing of 6. Atoms are represented by ellipsoids at the 50% probability level. Hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (deg): RuC(1), 2.0142(17); RuCl(1), 2.4212(4); RuPhCenter, 1.730; RuC(19), 2.0693(17); C(1)RuPhCenter, 129.9; C(19)RuPhCenter, 129.7; Cl(1)Ru(1)PhCenter, 126.1; C(1)RuCl(1), 92.29(5); C(19)RuCl(1), 86.04(5).
for over a week at 40 oC did not yield complex 6, as 5 is thermally stable. However, in presence of 1 equiv. of PCy3, 5 was transformed into 6 quantitatively at room temperature after 3 days (Scheme 2). PCy3 likely acts as a base to receive the HCl eliminated from 5 to generate HPCy3+Cl- which is observable by 1H, 31P NMR, and HRMS.
Scheme 2. PCy3 assisted CH insertion.
CD2Cl2 RT, 36 h
PCy3
> 95% Ru
Cl
Cl
N N
Ru Cl
N N
+ HPCy3+Cl
Scheme 3. A proposed mechanism. Ru PCy3 Ph Cl Cl
N N PCy3
+ PCy3 Ru
Ph Cl
Cl
N N CH insertion
Ru Cl Cl N N H -H insertion Ru Cl Cl N N reductive elimination Ru Cl Cl N N CH insertion
HPCy3+Cl- PCy3
Ru Cl
N N
3 7 8
6 5 9
Conclusion
We have reported the benzylidene insertion into an ortho CH bond of a phenyl group of an NHC ligand in an olefin metathesis catalyst 3. Further CH activation occurred by the assistance of the dissociated phosphine. These observations suggest that phenyl groups instead of mesityl groups of NHC-based ruthenium olefin metathesis catalysts are more vulnerable to decomposition via CH activation. New ligand design and synthesis of olefin metathesis catalysts are currently in progress to prevent this decomposition pathway while maintaining activity for tetrasubstituted olefin synthesis.
Experimental
Vacuum Atmospheres dry box (O2 < 2.5 ppm). NMR spectra were recorded on a Varian Inova (499.85 MHz for 1H; 202.34 MHz for 31P; 125.69 MHz for 13C) or on a Varian Mercury 300 (299.817 MHz for 1H; 121.39 MHz for 31P; 74.45 MHz for 13C). 31P NMR spectra were referenced using H3PO4 ( = 0 ppm) as an external standard. Elemental analyses were performed at Desert Analytics (Tucson, AZ). Mass spectra were recorded on JEOL JMS 600H spectrophotometer. Benzene, benzene-d6, and methylene chloride were dried by passage through solvent purification columns. CD2Cl2 was dried by vacuum transfer from CaH2. All solvents are degassed by either a generous Ar sparge or three freeze-pump-thaw cycles. (BIPh)(PCy3)(Cl)2Ru=(H)Ph (2) wasprepared according to literature procedure.9
Procedure for a typical decomposition measurement. 13.1 mg (0.0161 mmol) of complex 3 and ~ 1.0 mg (0.00561 mmol) of anthracene were weighed into a 1-dram vial. 0.700 mL of benzene-d6 or CD2Cl2 was used to transfer the sample to a screw-cap NMR tube. A screw-cap was used to seal the NMR tube, and this seal was reinforced with parafilm. The sample was placed into the spectrometer and allowed to equilibrate at the probe temperature for 10 minutes. Complex decomposition was following by monitoring the diminution of the benzylidene proton through collection of a time-delayed array of 1H NMR spectra (referred to as a preacquisition delay, PAD, by Varian software). Conversions to complex 5 and 6 were measured by monitoring characteristic 1H peaks of 6-bound phenyl group which show up in the region of 4.56.0 ppm.
solid was observed after 2 h. After 72 h, the precipitates were filtered, washed with benzene and dried under vacuum to afford complex 5 (19.0 mg, 0.0357 mmol, 58%). 1H NMR (CD2Cl2): 7.067.64 (m, 13H), 5.88 (dd, J = 6.0, 5.7 Hz, 1H), 5.80 (dd, J = 6.6, 5.4 Hz), 5.30 (d, J = 5.4 Hz, 1H), 4.92 (dd, J = 6.0, 5.1 Hz), 4.64 (d, J = 5.4 Hz, 1H), 3.93 (d, J = 14.5, 1H), 3.40 (d, J = 14.5, 1H). 13C{1H} NMR (CD2Cl2): 189.0, 138.8, 137.8, 137.7, 136.5, 134.3, 131.7, 131.5, 131.1, 129.4, 129.3, 129.2, 129.0, 128.9, 127.3, 124.6, 124.4, 111.7, 111.6, 100.4, 97.7, 93.9, 86.1, 85.7, 81.7, 36.4. HRMS analysis (FAB) m/z: Calcd [M+] 532.0047, found 532.0048.
Table 2. Summary of crystallographic data for 5 and 6.
5 6
formula C26H20Cl2N2Ru • 2(CH2Cl2) C26H19ClN2Ru • CH2Cl2
Mr 702.26 580.88
crystal color red/orange yellow
crystal size (mm) 0.38 x 0.28 x 0.15 0.30 x 0.26 x 0.07
crystal system monoclinic monoclinic
space group P21/c P21/n
a (Å) 9.5097(3) 13.8444(5)
b (Å) 12.7335(4) 13.1657(5)
c (Å) 24.0073(8) 14.0413(5)
(deg) 90 90
(deg) 99.6940(10) 117.6790(10)
(deg) 90 90
V (Å3) 2865.58(16) 2266.45(14)
Z 4 4
Dcalcd (g cm-3) 1.628 1.702
T (K) 200(2) 100(2)
(Å) 0.71073 0.71073
(mm-1) 1.128 1.065
R1a(all data) 0.0895 0.0862
wR2b (all data) 0.0798 0.0803
GOF 1.415 1.026
a
References and Notes
(1) Bourissou, D.; Guerret, O.; Gabbai, F. P.; Bertrand, G. Chem. Rev. 2000, 100, 3991. (2) Crudden, C. M.; Allen, D. P. Coord. Chem. Rev. 2004, 248, 22472273.
(3) Scott, N. M.; Nolan, S. P. Eur. J. Inorg. Chem. 2005, 18151828.
(4) Jazzar, R. F. R.; Macgregor, S. A.; Mahon, M. F.; Richards, S. P.; Whittlesey, M. K. J. Am. Chem. Soc. 2002, 124, 49444945.
(5) Dorta, R.; Stevens, E. D.; Nolan, S. P. J. Am. Chem. Soc. 2004, 126, 50545055. (6) Grundemann, S.; Kovacevic, A.; Albrecht, M.; Faller, J. W.; Crabtree, R. H. J. Am.
Chem. Soc. 2002, 124, 1047310481.
(7) Lebel, H.; Janes, M. K.; Charette, A. B.; Nolan, S. P. J. Am. Chem. Soc. 2004, 126, 50465047.
(8) Waltman, A. W.; Ritter, T.; Grubbs, R. H. Organometallics 2006, 25, 42384239. (9) Berlin, J. M.; Campbell, K.; Ritter, T.; Funk, T. W.; Chlenov, A.; Grubbs, R. H. Org.
Lett. 2007, 9, 13391342.
(10) The conversion of the RCM of diethyl dimethallylmalonate for catalyst 3 is 76% at room temperature after 24 h (vs. >95% for catalyst 3 at 40 oC, 24 h).
(11) Hong, S. H.; Day, M. W.; Grubbs, R. H. J. Am. Chem. Soc. 2004, 126, 74147415. (12) Hong, S. H.; Wenzel, A. G.; Salguero, T. T.; Day, M. W.; Grubbs, R. H. J. Am.
Chem. Soc. 2007, 129, in press.
(13) Ulman, M.; Grubbs, R. H. J. Org. Chem. 1999, 64, 72027207.
(15) Trnka, T. M.; Morgan, J. P.; Sanford, M. S.; Wilhelm, T. E.; Scholl, M.; Choi, T. L.; Ding, S.; Day, M. W.; Grubbs, R. H. J. Am. Chem. Soc. 2003, 125, 25462558. (16) Giunta, D.; Hölscher, M.; Lehmann, C. W.; Mynott, R.; Wirtz, C.; Leitner, W. Adv.
Synth. Catal. 2003, 345, 11391145.
(17) Abdur-Rashid, K.; Fedorkiw, T.; Lough, A. J.; Morris, R. H. Organometallics 2004, 23, 8694.
Chapter 5
Prevention of Undesirable Olefin Isomerization during Olefin Metathesis
Abstract
1,4-Benzoquinones have been found to prevent olefin isomerization of a number of allylic ethers and long-chain aliphatic alkenes during ruthenium-catalyzed olefin metathesis reactions. Electron-deficient benzoquinones are the most effective additives for the prevention of olefin migration. This mild, inexpensive, and effective method to block olefin isomerization increases the synthetic utility of olefin metathesis via improvement of overall product yield and purity.
Introduction
Olefin isomerization/migration is one of the side reactions in olefin metathesis that can significantly alter the product distribution and decrease the yield of the desired product, especially with ill-defined catalyst systems.1 Additionally, the side products resulting from unwanted isomerization are frequently difficult to remove via standard purification techniques. Well-defined ruthenium-based olefin metathesis catalysts such as 13 are generally highly selective for olefin metathesis. However, there have been some reports of
The majority of this chapter has been published: (a) Hong, S. H.; Sanders, D. P.; Lee, C.
olefin isomerization as well when the catalysts are stressed by high temperatures, high dilution and forced high turnovers.2-5
Ru PCy3 PCy3 Ph Cl Cl Ru Ru C N N Mes Mes Cl Cl Cl H N N Ru PCy3 Ph Cl Cl N N Ru Cl O Cl N N
1 2 3 4
While the exact mechanism(s) responsible for this isomerization are unknown (metal-based hydride, -allyl, or other pathways),6-9 recent results indicate that ruthenium hydride species such as 4 formed from the decomposition of the ruthenium metathesis catalysts can catalyze the migration of olefins under metathesis condition.10 This information has prompted us to develop a way to suppress the unwanted olefin isomerization reactions catalyzed by these metal hydrides.
Results and Discussion
Self-metathesis and isomerization of (Z)-5-t-butyldimethylsilyloxy-2-pentenoate 5 to the E-isomer 6 and silyl enol ether 7 served as an excellent system for initially studying the effects of additives on isomerization process.11 Compounds 5, 6, and 7 all have clearly distinguishable 1H NMR resonances. Through examination of the resultant E/Z ratio of the ,
(Table 1). Tricyclohexylphosphine oxide is an additive that has been reported to prevent isomerization of a specific substrate in a RCM reaction,7 however, it did not prevent the isomerization of 5 or the other substrates we tested. Acetic acid and 1,4-benzoquinone did not reduce the catalyst activity. Most metathesis reactions we tested were completed within an hour in presence of effective additives (Table 1, 2, and 3, and Scheme 2).12 However, we elongated the reaction time to 24 h to stress the catalysts to optimize isomerization.
Table 1. Self-metathesis reaction of (Z)-5-t-butyldimethylsilyloxy-2-pentenoate
OMe O
OTBS CD2Cl2, 40
oC, 24 h
MeO O
OTBS
+ OMe
O
OTBS
2 mol% 2, Additive
5 6 7
Product Distributiona
Additives Equiv.
(rel. to 5) 6+5 7b
None None 19%c 81%
2,2,2-Trifluoroethanol 1 11%c 89%
Hexafluoro-t-butyl alcohol 1 19%c 81%
Phenol 1 17%c 83%
Acetic Acid 0.1 >95%c None
Tricyclohexylphosphine oxide 0.1 22%c 78%
Maleic anhydride 1 >95%d None
1,4-Benzoquinone 0.1 >95%c None
a