RESEARCH
Improving the thermostability of a
fungal GH11 xylanase via site-directed
mutagenesis guided by sequence and structural
analysis
Nanyu Han
1,2†, Huabiao Miao
1†, Junmei Ding
1,2, Junjun Li
1,2, Yuelin Mu
1,2, Junpei Zhou
1,2and Zunxi Huang
1,2*Abstract
Background: Xylanases have been widely employed in many industrial processes, and thermophilic xylanases are in great demand for meeting the high-temperature requirements of biotechnological treatments. In this work, we aim to improve the thermostability of XynCDBFV, a glycoside hydrolase (GH) family 11 xylanase from the ruminal fun-gus Neocallimastix patriciarum, by site-directed mutagenesis. We report favorable mutations at the C-terminus from B-factor comparison and multiple sequence alignment.
Results: C-terminal residues 207-NGGA-210 in XynCDBFV were discovered to exhibit pronounced flexibility based on comparison of normalized B-factors. Multiple sequence alignment revealed that beneficial residues 207-SSGS-210 are highly conserved in GH11 xylanases. Thus, a recombinant xylanase, Xyn-MUT, was constructed by substituting three residues (N207S, G208S, A210S) at the C-terminus of XynCDBFV. Xyn-MUT exhibited higher thermostability than XynCDBFV at ≥70 °C. Xyn-MUT showed promising improvement in residual activity with a thermal retention of 14% compared to that of XynCDBFV after 1 h incubation at 80 °C; Xyn-MUT maintained around 50% of the maximal activity after incubation at 95 °C for 1 h. Kinetic measurements showed that the recombinant Xyn-MUT had greater kinetic efficiency than XynCDBFV (Km, 0.22 and 0.59 µM, respectively). Catalytic efficiency values (kcat/Km) of Xyn-MUT also increased (1.64-fold) compared to that of XynCDBFV. Molecular dynamics simulations were performed to explore the improved catalytic efficiency and thermostability: (1) the substrate-binding cleft of Xyn-MUT prefers to open to a larger extent to allow substrate access to the active site residues, and (2) hydrogen bond pairs S208-N205 and S210-A55 in Xyn-MUT contribute significantly to the improved thermostability. In addition, three xylanases with single point mutations were tested, and temperature assays verified that the substituted residues S208 and S210 give rise to the improved thermostability.
Conclusions: This is the first report for GH11 recombinant with improved thermostability based on C-terminus replacement. The resulting Xyn-MUT will be an attractive candidate for industrial applications.
Keywords: Xylanase, Thermostability, B-factor, MD simulation, Site-directed mutagenesis, C-terminus replacement
© The Author(s) 2017. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/ publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
Background
Xylan is the primary hemicellulosic constituent of plant cell walls and accounts for up to 35% of all renewable
organic carbon sources on Earth [1]. Xylan is a hetero-geneous polysaccharide consisting of a backbone chain of β-1,4-d-linked xylose units usually decorated with side groups (such as methyl and acetyl groups) and other sugar molecules [2]. Owing to the heterogeneity and complexity of xylan, complete degradation requires a set of main chain- and side group-cleaving enzymes [3]. Among all the xylanolytic enzymes, endo-β-1,4-xylanase
Open Access
*Correspondence: [email protected]
†Nanyu Han and Huabiao Miao contributed equally to this work 1 School of Life Sciences, Yunnan Normal University, Kunming 650500,
China
(xylanase; EC 3.2.1.8) is a pivotal enzyme that is capable of randomly hydrolyzing the internal β-1,4-d-xylosidic linkages in the backbone chain to yield xylooligosaccha-rides (XOSs) of various chain lengths during xylan bio-degradation [2, 4].
Xylanases have been widely applied in industry, includ-ing paper and pulp processinclud-ing, feed manufacture, and next generation biorefineries [2, 5–7]. Due to the harsh conditions of biotechnological treatments, xylanases with advantageous properties such as excellent thermostabil-ity, broad pH adaptabilthermostabil-ity, and high specific activity are in high demand [8]. As such, numerous projects have been undertaken to discover and develop novel xylanases with favorable properties [9–11].
Based on catalytic domain sequence similarity, xyla-nases from various source organisms are classified into glycoside hydrolase (GH) families 5, 8, 10, 11, 30, and 43 by the CAZy database, and principally belong to GH10 and GH11 [12]. In contrast to GH10 xylanases, GH11 xylanases display higher catalytic efficiency, higher sub-strate selectivity, and a greater variety of temperature and pH optima [13]. These advantageous properties make GH11 more suited for industrial applications. Thus, the determinants for the improved properties of GH11 have been widely explored [13–17]. Among them, understand-ing the heat-resistance mechanism for GH11 has become an intense research area owing to the high-temperature requirement in various industrial conditions.
It has been widely accepted that N-terminal region (NTR) of GH11 is of great importance in maintain-ing xylanase thermostability, and several heat-resistant recombinants have been developed by replacing the NTR with the corresponding parts from thermostable xyla-nases [17–19]. In 2014, the Rey-Ting Guo group solved the crystal structure of a GH11 xylanase (XynCDBFV) from the ruminal fungus Neocallimastix patriciarum [20]. XynCDBFV was identified to have the longest NTR among all GH11 members [20]. The NTR of XynCDBFV folds into an α-helix and tightly attaches to a β-sheet via a disulfide bridge (DS1). It has been shown that the NTR plays an important role in XynCDBFV thermosta-bility and that DS1 is the critical factor joining the NTR to the main body of XynCDBFV [20]. Additionally, one thermophilic xylanase (NFX) from Nonomuraea flexu-osa has been previously reported and crystallized. This thermophilic NFX was stable at 80 °C and even retained partial activity at 90–100 °C [21]. Although there were no evident structural reasons for the significant higher ther-mostability of NFX, an additional GNPGNP sequence was found at the C-terminus [21]. Therefore, the C-ter-minus of NFX and other thermophilic xylanases may also play a pivotal role in maintaining thermostability. A thermophilic xylanase recombinant may be constructed
by combining crucial thermostability-enhancing compo-nents of xylanases, such as the NTR from XynCDBFV, C-terminus from NFX.
In this work, the sequence and structure of XynCDBFV were carefully compared to those of thermophilic xyla-nases including NFX. C-terminal residues 207-SSGS-210 were found to be highly conserved in thermophilic xyla-nases. However, residues 207–210 with sequence NGGA of XynCDBFV were found to display pronounced flexibil-ity. Subsequently, three residues, N207, G208, and A210, from 207-NGGA-210 of XynCDBFV were substituted with serines by site-directed mutagenesis. Experimental measurements showed that the triple mutant displayed higher thermostability and catalytic efficiency than wild-type XynCDBFV. Moreover, heat-resistance mechanisms for the triple mutant were explored by molecular dynam-ics (MD) simulations and verified by single point muta-tions. To our knowledge, this is the first report of a GH11 recombinant with improved thermostability based on C-terminus replacement, and the triple mutant demon-strates attractive properties for industrial use.
Results
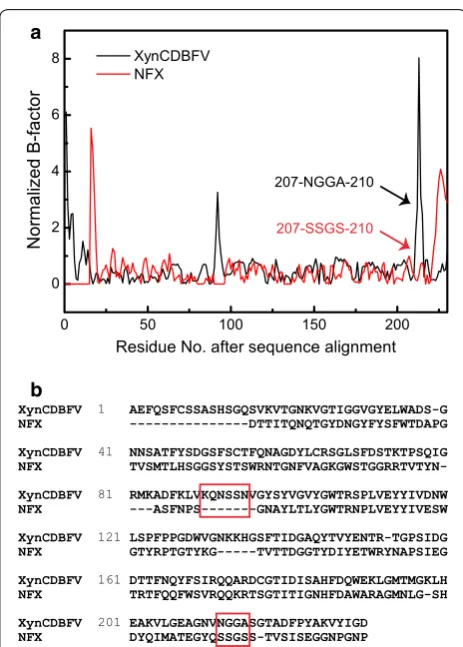
Mutagenesis sites predicted by B‑factor comparison B-factors determined from X-ray diffractions are lin-early related to the mean square displacement of atoms relative to their average positions [22]. Therefore, crys-tal structure B-factors provide useful information about protein dynamics, structural flexibility, and protein sta-bility [23]. In this work, the B-factors of XynCDBFV and NFX were extracted from the crystal structures. After pairwise sequence alignment, normalized B-factors for each Cα atom were compared between XynCDBFV and NFX (Fig. 1a). Two segments corresponding to pro-nounced flexibility in XynCDBFV were discovered: one segment from residue K86 to residue N91 and another from residue N207 to residue A210. Residues K86 to N91 in XynCDBFV correspond to sequence gaps in NFX (Fig. 1b). Residues N207 to A210 (207-NGGA-210) in XynCDBFV correspond to residues 207-SSGS-210 in NFX and are located at the C-terminal region. It has been suggested that the C-terminus plays a crucial role in maintaining NFX thermostability. Thus, we focused on the second segment of residues N207 to A210.
xylanase is highly related with the arrangement and com-pactness of β-sheets. Analogous residues 207–210 with different secondary structures may contribute to various stabilities among GH11xylanases.
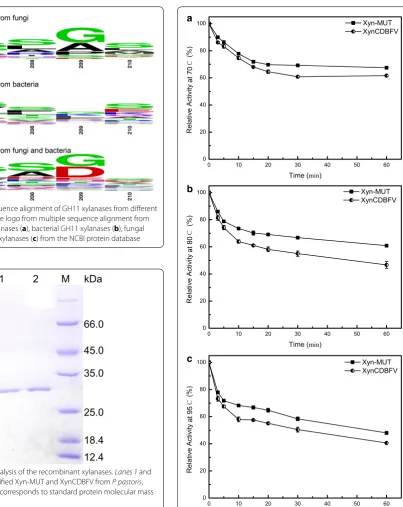
B-factor and structural comparisons indicate that resi-dues 207–210 potentially influence xylanase stability. XynCDBFV is a fungal xylanase from N. patriciarum and NFX is a bacterial xylanase from N. flexuosa. To investi-gate sequence diversity of residues 207–210 in different organisms, all GH11 xylanase sequences from fungi and bacteria in the NCBI database were compared (Fig. 3). Multiple sequence alignment revealed that beneficial res-idues S207, S208, G209, and S210 are highly conserved in both fungal and bacterial GH11 xylanases (Fig. 3a, b). Specifically, residues S207, S208, G209, and S210 account for 31, 31, 48, and 31% in fungal GH11 xylanases, respec-tively. While the sequence combination of 207-NGGA-210 is much less conserved: N207, G208, G209, and A207-NGGA-210 only account for 7, 4, 48, and 11% in fungal GH11 xyla-nases, respectively. Normalized B-factor comparison, plus multiple sequence alignment suggests 207-SSGS-210 is a putative segment in improving GH11 xylanase thermostability.
Construction and characterization of Xyn‑MUT
Inspired by the B-factor comparison and multi-ple sequence alignment, residues 207-NGGA-210 in XynCDBFV were substituted to 207-SSGS-210 at equiva-lent sites by site-directed mutagenesis. The resultant tri-ple mutant (N207S, G208S, A210S) is called Xyn-MUT in this work. Xyn-MUT displayed the same molecular mass (27.37 kDa) as XynCDBFV on SDS-PAGE (Fig. 4). The specific activities of purified Xyn-MUT and XynCDBFV were 920.17 and 798.34 U/mg, respectively.
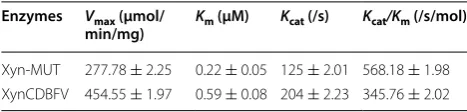
To evaluate thermostability, residual activities of Xyn-MUT and XynCDBFV were measured after incuba-tion at various temperatures for 1 h. Both Xyn-MUT and XynCDBFV were stable at 70 °C. Residual activi-ties for Xyn-MUT and XynCDBFV were 67 and 62% after 1 h treatment, respectively (Fig. 5a). At tempera-ture >70 °C, Xyn-MUT showed greater residual activity
207-NGGA-210
207-SSGS-210
XynCDBFV 1 AEFQSFCSSASHSGQSVKVTGNKVGTIGGVGYELWADS-G
NFX ---DTTITQNQTGYDNGYFYSFWTDAPG
XynCDBFV 41 NNSATFYSDGSFSCTFQNAGDYLCRSGLSFDSTKTPSQIG
NFX
TVSMTLHSGGSYSTSWRNTGNFVAGKGWSTGGRRTVTYN-XynCDBFV 81 RMKADFKLVKQNSSNVGYSYVGVYGWTRSPLVEYYIVDNW
NFX ---ASFNPS---GNAYLTLYGWTRNPLVEYYIVESW
XynCDBFV 121 LSPFPPGDWVGNKKHGSFTIDGAQYTVYENTR-TGPSIDG
NFX GTYRPTGTYKG---TVTTDGGTYDIYETWRYNAPSIEG
XynCDBFV 161 DTTFNQYFSIRQQARDCGTIDISAHFDQWEKLGMTMGKLH
NFX TRTFQQFWSVRQQKRTSGTITIGNHFDAWARAGMNLG-SH
XynCDBFV 201 EAKVLGEAGNVNGGASGTADFPYAKVYIGD
NFX DYQIMATEGYQSSGSS-TVSISEGGNPGNP
a
b
Fig. 1 Comparison of normalized B-factors after pairwise sequence alignment. a Normalized B-factors were compared between XynCD-BFV (black) and NFX (red). b Pairwise sequence alignment between XynCDBFV and NFX
a b
than XynCDBFV. Xyn-MUT retained 61% activity after 1 h incubation at 80 °C, while XynCDBFV retained no greater than 50% activity (47%) at the same condition (Fig. 5b). Moreover, the residual activity for Xyn-MUT was approximately 50% after incubation at 95 °C for 1 h, while the retained activity for XynCDBFV was 40% at the same condition (Fig. 5c). These results highlight that the three substitutions (N207S, G208S, A210S) are advanta-geous for GH11 xylanase thermostability.
Kinetic Analysis of Xyn‑MUT and XynCDBFV
Kinetic parameters were determined at 37 °C for XynCD-BFV and Xyn-MUT. Reactions were monitored using the DNS method at eight concentrations of beechwood xylan, from 0.5 to 5 mg/mL. Kinetic measurements Fig. 3 Multiple sequence alignment of GH11 xylanases from different
organisms. Sequence logo from multiple sequence alignment from all fungal GH 11 xylanases (a), bacterial GH11 xylanases (b), fungal and bacterial GH11 xylanases (c) from the NCBI protein database
Fig. 4 SDS-PAGE analysis of the recombinant xylanases. Lanes 1 and 2 correspond to purified Xyn-MUT and XynCDBFV from P. pastoris, respectively; lane M corresponds to standard protein molecular mass markers
a
b
c
showed that the apparent Km values for Xyn-MUT
and XynCDBFV were 0.22 and 0.59 μM, respectively (Table 1). The smaller Michaelis constant (Km) of
Xyn-MUT indicates an increase in kinetic efficiency compared to that of XynCDBFV. Moreover, catalytic efficiency (kcat/
Km) of Xyn-MUT was also increased (1.64-fold). Kinetic
analysis revealed that the three substituted residues, although primarily designed to improve thermostability, also enhanced catalytic efficiency and substrate binding.
Stability of mutagenesis sites in MD simulations
To gain insight into the improved thermostability and catalytic efficiency of XynCDBFV, MD simulations for XynCDBFV and Xyn-MUT at different temperatures (65/80 °C) were conducted. The root mean square fluc-tuation (RMSF) reflects the flexibility for each residue during simulations. Comparing the RMSF values of the mutated residues at 65 and 80 °C in the two xylanases (Table 2), we found that RMSF values of most mutated residues at 80 °C were higher than those at 65 °C in both XynCDBFV and Xyn-MUT, suggesting enhanced flexibility of the mutated residues at elevated tempera-ture. Specifically, mutant residue S207 in Xyn-MUT has smaller RMSF values than N207 in XynCDBFV at both 65 and 80 °C. The Xyn-MUT S208 RMSF (0.1358 nm) was smaller than that of XynCDBFV G208 (0.1524 nm) at 65 °C; however, the situation is reversed at 80 °C, with the Xyn-MUT S208 RMSF (0.1917 nm) larger than that of XynCDBFV G208 (0.1653 nm). The RMSF values of Xyn-MUT S210 and XynCDBFV A210 were almost the same at 65 °C. At 80 °C, the RMSF of Xyn-MUT S210 (0.0769 nm) was smaller than that of XynCDBFV A210 (0.0863 nm). Although the RMSF discrepancies were not as significant as that of the crystallographic B-factors, the smaller RMSF values of Xyn-MUT indicate the mutated sites were relatively stable during simulation.
Comparison of the substrate‑binding cleft between Xyn‑MUT and XynCDBFV
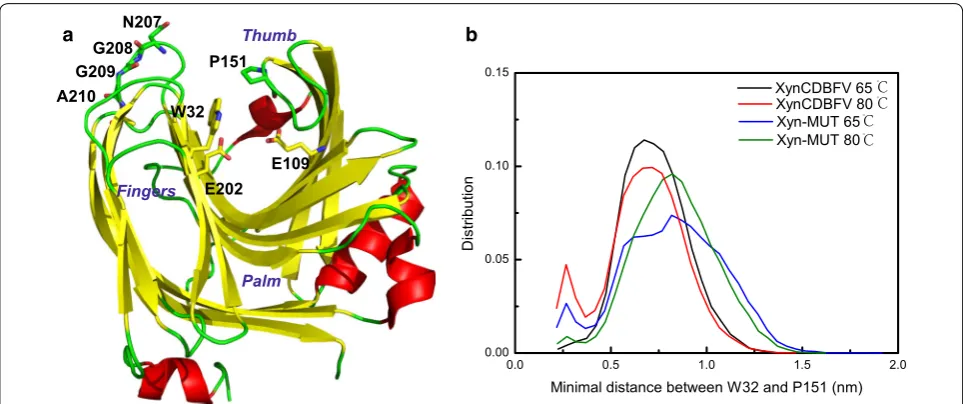
The β-jelly-roll catalytic domain of xylanase resembles a partially closed right hand and is made of two anti-paral-lel β-sheets sculpting a long and deep substrate-binding cleft. The β-sheets form the “palm and fingers” and one long loop forms the “thumb”, which partially closes the cleft. The catalytic residues in XynCDBFV and Xyn-MUT are the same (E109 and E202), and are located at the palm
and fingers side of the cleft (Fig. 6a). The thumb loop is highly conserved in GH11 xylanases and is the most flex-ible region based on crystal structure comparison [24]. It has been suggested that the elevated flexibility of the thumb loop is important to allowing substrate access to the active site. The narrowest zone of the cleft is localized between the conserved proline (P151) in the thumb loop and the conserved tyrosine/tryptophan (W32) in the fin-gers domain [13]. In order to understand the open and closed states of the substrate-binding cleft in XynCDBFV and Xyn-MUT, the minimal distance between W32 and P151 was calculated each entire simulation trajectory. Comparing the minimal distance (W32–P151) distri-bution between XynCDBFV and Xyn-MUT (Fig. 6b), it is obvious that the substrate-binding cleft in Xyn-MUT prefers to open to a larger extent at both 65 and 80 °C with the minimal distance centered around 0.8 nm. On the other hand, the minimal distance between W32 and P151 in XynCDBFV at the two temperatures centered at 0.7 nm, indicating a narrower gate to the substrate-binding cleft compared to that of Xyn-MUT. The larger opening of the substrate-binding cleft in Xyn-MUT may contribute to the higher kinetic efficiency of Xyn-MUT.
Additionally, solvent-accessible surface areas for the catalytic residues E109 and E202 were calculated along the whole trajectories (Table 3). The accessible areas of E109 in XynCDBFV and Xyn-MUT were almost the same at both temperatures. Intriguingly, the accessible area of E202 in Xyn-MUT was larger than that of E202 in XynCDBFV at both 65 and 80 °C, indicating that E202 in Xyn-MUT has higher probability of contacting the sub-strate. The greater W32–P151 minimal distance distribu-tion and greater E202 accessible area of Xyn-MUT versus XynCDBFV provide a molecular-level explanation for the higher kinetic rate and catalytic efficiency.
Improved thermostability explored by MD simulations and verified by single point mutations
Generally, mutations that improve thermostability may result via formation of hydrogen bonds, disulfide bridges, stabilization of β-turns or flexible terminuses, enhance-ment of hydrophobic packing, or α-helix or β-sheet sta-bility [25]. In our work, residues 207–210 were previously Table 1 Kinetics of Xyn-MUT and XynCDBFV
Enzymes Vmax (μmol/
min/mg) Km (μM) Kcat (/s) Kcat/Km (/s/mol)
Xyn-MUT 277.78 ± 2.25 0.22 ± 0.05 125 ± 2.01 568.18 ± 1.98 XynCDBFV 454.55 ± 1.97 0.59 ± 0.08 204 ± 2.23 345.76 ± 2.02
Table 2 RMSF for mutated residues over the whole simula-tion in XynCDBFV and Xyn-MUT at 65 and 80 °C
Enzymes N/S207 (nm) G/S208 (nm) A/S210 (nm)
discovered to form a β-turn and β-strand in XynCDBFV and NFX crystal structures, respectively (Fig. 2). It is well known that β-strands are connected laterally by hydro-gen bonds between backbone chains [26], so we focused on analyzing β-sheet stability and hydrogen bonds inter-actions with the mutated sites.
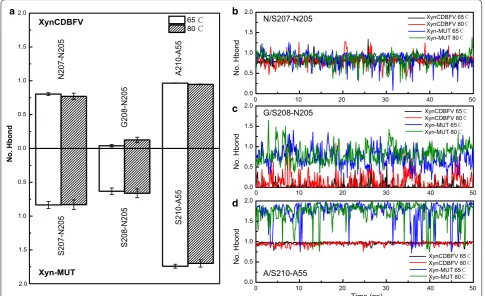
Hydrogen bonds interactions around the C-terminal sites in XynCDBFV and Xyn-MUT were monitored along the whole trajectories (Fig. 7). In XynCDBFV, the prob-ability of hydrogen bonding between N207 and N205 slightly decreased at 80 °C compared to that at 65 °C (Fig. 7a). However, in Xyn-MUT the hydrogen bond connecting S207 and N205 was well preserved at high temperature (80 °C). There was no significant difference in number of hydrogen bonds formed between N/S207 and N205 in XynCDBFV and Xyn-MUT based on time evolution analysis (Fig. 7b). The hydrogen bond con-necting S208 and N205 in Xyn-MUT was much stronger (>30%) than that between G208 and N205 in XynCDBFV (Fig. 7a), and the hydrogen bond connecting S208 and
N205 in Xyn-MUT was well preserved during the whole simulation (Fig. 7c). S210 and A55 in Xyn-MUT dis-played a moderately loose interaction at 80 °C; however, the number of hydrogen bonds between S210 and A55 was 1.85-fold larger than that of XynCDBFV (Fig. 7a). In both XynCDBFV and Xyn-MUT, the backbone nitrogen of A210/S210 forms one stable hydrogen bond with oxy-gen in the backbone carboxyl group of A55. Moreover, the oxygen (OG) in the side-chain hydroxyl group of S210 in Xyn-MUT has more than 80% probability of forming a hydrogen bond with the oxygen (O) in the backbone carboxyl group of A55 based on structural analysis. The additional hydrogen bond between S210 (OG) and A55 (O) was well maintained during the simulation (Fig. 7d). Thus, the increased number of hydrogen bonds between mutated residue S210 and A55 in Xyn-MUT may be a dominant reason for the improved thermostability. In brief, mutations to S208 and S210 may give rise to the improved thermostability of Xyn-MUT.
Furthermore, the contribution of each individual mutated residue to Xyn-MUT’s thermostability was veri-fied by comparing XynCDBFV to the three single xyla-nase mutants (N207S, G208S, and A210S) expressed in Escherichia coli (Additional file 1: Figure S1). Tem-perature assays revealed that two single mutants, G208S and A210S, exhibited higher residual activity at 65 °C (Fig. 8). G208S and A210S showed improvements in residual activity with greater thermal retention of 4 and 5%, respectively, compared to XynCDBFV after incuba-tion at 65 °C for 20 min. In contrast, the residual activi-ties of N207S were lower than that of XynCDBFV when
a b
Fig. 6 Open and closed states of substrate-binding sites in two xylanases. a Schematic structure of XynCDBFV colored by secondary structure, β-strand in yellow, α-helix in red and loop in green. b Minimal distance distribution between W32 and P151 in XynCDBFV and Xyn-MUT at 65 and 80 °C
Table 3 Solvent-accessible surface area for catalytic resi-dues (E109 and E202) over the whole simulation in XynCD-BFV and Xyn-MUT at 65 and 80 °C
Enzymes E109 (nm2) E202 (nm2)
incubated at 65 °C, indicating that introducing the N207S substitution did not contribute to higher Xyn-MUT thermostability. The experimental results were in good
agreement with the MD simulations, both indicating that the mutated residues S208 and S210 predominantly con-tribute to the improved thermostability of Xyn-MUT.
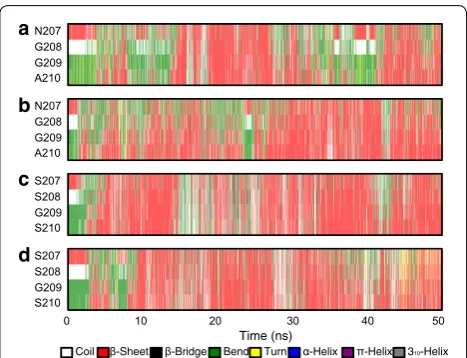
To decipher structural alterations of the mutated sites during the simulations, secondary structure propen-sity was analyzed by DSSP [27]. The time evolution of the secondary structure demonstrated the process of conformational conversion (Fig. 9). It is apparent that N207 and G208 in XynCDBFV were associated with a lower propensity for forming a β-sheet than Xyn-MUT (sparse red lines in Fig. 9a, b). Intriguingly, although both XynCDBFV and Xyn-MUT have a glycine at position 209, the propensity of G209 forming a β-sheet was 36% in XynCDBFV at 65 °C (Fig. 9a), but 59% in Xyn-MUT (Fig. 9c). This may be the influence of local secondary structure formation: segment 207-SSGS-210 in Xyn-MUT had a higher propensity for forming a β-sheet than that in XynCDBFV, so G209 in Xyn-MUT’s middle seg-ment would automatically be inclined to take a β-sheet conformation. Moreover, S210 in Xyn-MUT had higher propensity for forming a β-sheet than A210 in XynCD-BFV: S210 had 57 and 50% propensities to form a β-sheet in Xyn-MUT at 65 and 80 °C, respectively (Fig. 9c, d);
a b
c
d
Fig. 7 Statistics of hydrogen bond interactions with substituted residues in two xylanases. a Number of hydrogen bonds interacted with substi-tuted residues in XynCDBFV (upper) and Xyn-MUT (lower) over the whole simulations. Number of hydrogen bonds in three pairs N/S207-N205 (b), G/ S208-N205 (c), and A/S210-A55 (d) as a function of simulation time in XynCDBFV and Xyn-MUT at 65 and 80 °C
however, A210 had no more than 50% propensity for forming a β-sheet in XynCDBFV at both temperatures (Fig. 9a, b). Above all, secondary structure propensity analysis revealed that residues in 207-SSGS-210 of Xyn-MUT had a higher propensity for forming a β-sheet structure than 207-NGGA-210 in XynCDBFV.
Discussion
Developing a thermostable xylanase is of great value to meeting practical industrial demands. Additionally, enhanced temperature stability may also improve kinetic efficiency [28]. In this work, B-factor comparison and multiple sequence alignment were first performed to guide the design of mutations to obtain a thermostable xylanase. The result, Xyn-MUT, constructed by site-directed mutagenesis, showed higher thermostability and catalytic efficiency, and the heat-resistance mechanisms of Xyn-MUT were explored by MD simulations and sin-gle point mutations.
Firstly, the comparison of normalized B-factors between XynCDBFV and thermophilic NFX is the most crucial step in constructing a thermostable recombinant in this work. Systematic structural studies regarding various enzymes have demonstrated that thermophilic enzymes are characterized by higher degrees of rigidity [29]. Therefore, a strategy for enhancing the thermosta-bility of a particular enzyme is to increase its rigidity at appropriate sites. The B-factors from X-ray data pro-vide information on the fluctuation, and hence, rigid-ity of atoms relative to their equilibrium positions [22]. Previous work has successfully demonstrated improving
enzyme thermostability through the interpretation of B-factors [30]. Consequently, we calculated and com-pared normalized B-factor values from XynCDBFV and NFX, determined residues with pronounced degrees of flexibility, and constructed a recombinant Xyn-MUT by mutating the flexible residues. Temperature stability testing and kinetic analysis revealed that Xyn-MUT dis-plays higher thermostability and catalytic efficiency than XynCDBFV. Experimental measurements validate the accuracy of B-factor interpretation.
Secondly, Serine (Ser) at the C-terminus is highly related to thermostability in GH11 xylanase. Multiple sequence alignment showed that residues S207, S208, and S210 are highly conserved in both fungal and bac-terial GH11 xylanases. The recombinant Xyn-MUT was produced by substituting residues in position 207, 208, and 210 to Ser. The more thermostable Xyn-MUT indi-cates that incorporating Ser is a potent strategy in engi-neering a thermostable GH11 xylanase. In another study, Ser- and Thr-containing mutants displayed less flexibility in thermophiles than in mesophiles based on analysis of B-factors from mesophilic and thermophilic proteins [23], suggesting that Ser and Thr are associated with high rigidity in thermophiles. Accordingly, this approach to engineering a thermostable enzyme requires two steps: (1) determine appropriate sites with high flexibility; and (2) mutate flexible sites to appropriate amino acids that allow high rigidity. In our work, the first step was assisted by B-factor comparison. In the second step, the flexible residues were mutated to Ser, a residue that contributes to high rigidity in thermophiles.
Thirdly, this is the first report of a GH11 recombinant with improved thermostability based on C-terminal replacements. Until now, rational design of improved GH11 xylanases have typically focused on three aspects: (1) replacement of the NTR with corresponding parts from thermostable enzymes [17–19]; (2) stabilization of α-helices by introducing disulfide bridges or electrostatic interactions [21, 31]; (3) modification of surface charac-teristics to form a tighter packing enzyme with fewer cav-ities [16, 32]. In this work, we found a dominant sequence SSGS-210 at the C-terminus and replaced 207-NGGA-210 in XynCDBFV with this dominant sequence. The thermostable recombinant provides new opportuni-ties for engineering GH11 xylanases at the C-terminal region.
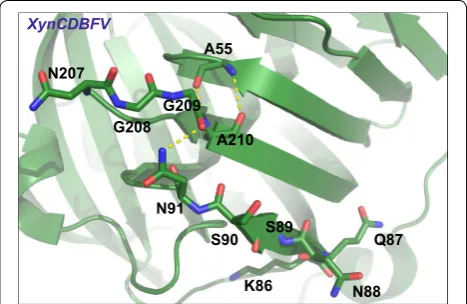
Fourthly, the first segment 86-KQNSSN-91 determined by B-factor comparison may also influence the stability of XynCDBFV. This segment corresponds to sequence gaps in NFX from pairwise sequence alignment. We did not construct XynCDBFV mutants that delete residues 86-KQNSSN-91 in this work. However, the residues 86-KQNSSN-91 are located adjacent to the β-strand a
b
c
d
formed by the second segment 207-NGGA-210 (Fig. 10). Moreover, N91 interacts with A210 through a hydro-gen bond. Residues 207-NGGA-210 displayed flexibility during MD simulations; the N91 interaction plus other residues in 86-KQNSSN-91 may also disturb the stabil-ity of XynCDBFV. This conjecture requires experimental studies.
Fifthly, the additional GNPGNP sequence at the C-ter-minus of NFX is associated with larger-than-average normalized B-factor values (Fig. 1a), indicating it may serve as a flexible linker at the C-terminus. This may allow relatively free movement of the C-terminal domain. Although the GNPGNP sequence from NFX may not contribute to thermostability, it may assist in maintaining NFX’s function.
Lastly and importantly, XynCDBFV and its mutants expressed in E. coli are less active than that expressed in Pichia pastoris. The E. coli expression system was used to test the thermostability contribution of individual mutated residues in isolation, since the E. coli expression system is easier to handle than the P. pastoris expression system. However, we discovered that XynCDBFV and its three single mutants expressed in E. coli exhibit their highest activities at no more than 65 °C. Similar phenom-ena have been observed in several studies [20, 33, 34], indicating that P. pastoris is a better production host than E. coli for XynCDBFV expression and performance, and a more suitable expression system for commercial appli-cations. Nevertheless, the enhanced thermostability of Xyn-MUT accounted for by replacing G208 and A210 in XynCDBFV with serines.
Conclusions
In this work, we identified for the first time the conserved C-terminal residues 207-SSGS-210 of GH11 xylanases and constructed a recombinant xylanase, Xyn-MUT.
Experiments showed Xyn-MUT had higher thermosta-bility and kinetic and catalytic efficiency than those of its template (XynCDBFV). Heat-resistance mechanisms explored by MD simulations and single point muta-tion studies revealed that S208 and S210 give rise to this improved thermostability. Additionally, residues 207-SSGS-210 assist Xyn-MUT in forming a more stable and compact β-sheet structure. The resulting Xyn-MUT is an attractive candidate in industrial applications. This work confirms that sequence/structural-guided protein engi-neering is an effective strategy for obtaining a thermosta-ble enzyme.
Methods Materials
High-fidelity DNA polymerase, restriction endonuclease (NdeI, XhoI), and dNTP were purchased from TaKaRa (Otsu, Japan). pEASY-E2 expression vector and pMD19-T vector were purchased from pMD19-TaKaRa (Otsu, Japan). pPICZA expression vector was purchased from Invitro-gen (Shanghai, China). The plasmid mini-prep kit and DNA gel extraction kit were purchased from Omega (Taipei, USA). One Step cloning kit was purchased from Vazyme biotech (Nanjing, China). Fast MultiSite Mutagenesis System and Bradford protein assay kit were purchased from TransGen (Beijing, China). Escherichia coli Trans1-T1 cells, E. coli BL21 (DE3) cells, and E. coli DMT cells were purchased from TransGen (Beijing, China). All other chemicals were of analytical grade and commercially available.
Gene cloning and site‑directed mutagenesis
XynCDBFV (GenBank:KP691331) was synthesized in Generay (Shanghai Generay Biotech Co. Ltd.). The gene of xylanase triple mutant (Xyn-MUT) and three xyla-nase single mutants (N207S, G208S, and A210S) were constructed by introducing mutations to XynCDBFV through site-directed mutagenesis. Mutations causing the amino acid exchanges were introduced using the Fast MultiSite Mutagenesis System according to the manu-facturer’s instructions. Forward and reverse primers for XynCDBFV, Xyn-MUT, and three single mutant genes are listed in Table 4. The XynCDBFV and Xyn-MUT PCR products were linked with pPICZA vector and then transformed into P. pastoris system; the resulting recom-binants containing XynCDBFV and Xyn-MUT were con-firmed by DNA sequencing. On the other hand, N207S, G208S, A210S, and XynCDBFV PCR products were linked with pEASY-E2 vector and then transformed into E. coli Trans1-T1 for sequencing. PCR cycling conditions consisted of an initial step of 5 min at 94 °C, followed by 30 cycles of 30 s at 94 °C, 1 min at 55 °C, and 3 min 30 s at 72 °C.
Protein expression and purification
Plasmids pPICZA/XynCDBFV and pPICZA/Xyn-MUT were linearized by PmeI and then individually trans-formed into P. pastoris GS115 by electroporation. The transformants were selected on YPD (yeast extract pep-tone dextrose) plates containing 200 μg/mL zeocin. The selected clones were inoculated and amplified in 30 mL of buffered glycerol-complex medium at 30 °C for 2 days. Then the culture medium was replaced by 20 mL of buffered methanol-complex medium to induce protein expression. For purification, the supernatants were con-centrated using Amicon centrifugal filter device (cutoff 3.000). On the other hand, E. coli BL21 (DE3), harboring pEASY-E2 vector that link XynCDBFV and three single mutant genes, was grown overnight at 37 °C in Luria– Bertani (LB) medium supplemented with 100 μg/mL ampicillin. Afterwards, the culture was transferred into fresh LB medium containing 100 μg/mL ampicillin by 1% dilution at 37 °C. IPTG (0.1 mM) was added until cell density (OD600nm) reached 0.6–0.8, then the culture was
grown at 22 °C for 12 h [35]. Cells were then harvested by centrifugation and resuspended with PBS buffer (pH 7.0, 140.0 mM NaCl, 2.7 mM KCl, 10.0 mM Na2HPO4,
1.8 mM KH2PO4). Supernatant was collected after the
cells were disrupted by sonication and centrifugation at 15,000 rpm for 30 min at 4 °C. The C-terminal His-tagged xylanases were purified using a column of Ni–NTA aga-rose. The success of the purification was determined by SDS-PAGE, and enzyme concentration was determined by Bradford protein assay kit.
Enzyme activity characterization
Xylanase activity was analyzed by measuring the release of reducing sugars (xylose) liberated from birchwood xylan using 3,5-dinitrosalicylic acid under optimum conditions [36]. One activity unit (1 U) was defined as the amount of xylanase required to liberate 1 μmol of reducing sugars per minute. All assays in this work were performed in triplicate. Thermostability was assayed by measuring residual enzyme activity after incubation at 70, 80, and 95 °C for 1 h under xylanase optimal pH. The residual enzyme activities were measured under standard assay.
The kinetic parameters (Km, Vmax, and kcat) for purified
xylanases were determined in McIlvaine buffer (pH 5.5) at 37 °C. Birchwood xylan was used as the substrate. The reactions were monitored using the DNS method at eight concentrations of beechwood xylan, from 0.5–5 mg/ mL. The kinetic values were calculated by fitting the Lineweaver–Burk plot [37].
Prediction of the mutagenesis sites by B‑factor comparison To evaluate the main chain flexibility, B-factors of the Cα atoms in XynCDBFV and NFX were extracted from the PDB files [20, 21]. Because the B-factors in different PDB files were refined in different procedures, they cannot be directly compared [38]. Thus, the B-factors in each pro-tein were normalized to have a distribution of zero mean and unit variance based on the following equation:
where the 〈B〉 is the average of all Cα atoms and σ(B) is the standard deviation of the B-factors for the individual protein [39]. The above equation has already been testi-fied and used by other groups [40, 41].
MD simulation details
The X-ray crystal structure of XynCDBFV was taken from PDB 3WP4 [20] and the recombinant with three point mutations (N207S, G208S, A210S), Xyn-MUT, was built by SWISS-MODEL sever [42]. Normal MD simula-tions of XynCDBFV and Xyn-MUT were performed at 65 and 80 °C. After 1000-step energy minimization, all the systems were first equilibrated for 5 ns in NPT ensemble followed by another 5 ns equilibration in NVT ensemble by restraining all heavy atoms. Finally, each system was simulated for 50 ns, and the simulation time for all the simulation systems was 200 ns in total. All systems were solvated with TIP3P waters in an octahedral box [43], and the minimal distance between each protein and edge of the box was set to 0.8 nm. Sodium and chloride ions were added with a concentration of 100 mM to neutral-ize the systems. Protonation states for histidines were
B′=
B− �B�
σ (B) ,
Table 4 Oligonucleotide primers for XynCDBFV, Xyn-MUT,
and three single mutant genes
Nucleotide resulting in the desired mutation is underlined and in bold
Primers Primer sequences
XynCDBFV-forward 5′-CAAAGTTTCTGTAGTTCAGCTTCTC-3′ XynCDBFV-reverse 5′-ACCAATGTAAACCTTTGCGTATG-3′ Xyn-MUT-1-forward 5′-GGTGAAGCCGGTAACGTTAGCA
GTGGTGC-CAGTG-3′
Xyn-MUT-1-reverse 5′-CACTGC TAACGTTACCGGCTTCACCTAAAAC-CTTG-3′
Xyn-MUT-2-forward 5′-GGTAACGTTAGCAGTGGTTCCAGTGGTAC-3′ Xyn-MUT-2-reverse 5′- A
ACCACTGCTAACGTTACCGGCTTCAC-CTAAAAC-3′
determined by the UCSF Chimera program [44]. The GROMACS program suite version 4.5.7 and Amber ff99SB force field were used in all simulations [45, 46]. The simulations were performed in an isothermal–iso-baric ensemble (65/80 °C, 1 bar). Bond length constraints were applied to all bonds that contained hydrogen atoms based on the LINCS protocol [47]. An integration step of 0.002 ps was used in all simulations. Electrostatic inte-grations were treated with Particle Mesh Ewald method with a cutoff of 0.9 nm with grid spacing for the FFT grid <0.12 nm [48].
Hydrogen bond analysis
Hydrogen bonds between mutational residues and nearby residues in all simulation systems were analyzed by using g_hbond in the GROMACS suite. Geometri-cal criterions which include donor–acceptor distance and hydrogen-donor–acceptor angle are used to calcu-late hydrogen bond. In Fig. 7a, the number of hydrogen bonds was calculated based on the whole 50 ns simu-lation in each system, and the error bar represents one standard error which was calculated based on the aver-aged number of hydrogen bonds every 10 ns in each sys-tem. In Fig. 7b–d, the hydrogen bond forming probability was analyzed every 100 ps during the whole simulation.
Abbreviations
DS1: disulfide bridge; E. coli: Escherichia coli; GH: glycoside hydrolase; LB: Luria– Bertani; MD: molecular dynamics; NTR: N-terminal region; P. pastoris: Pichia pastoris; XOSs: xylooligosaccharides.
Authors’ contributions
NH carried out the computational prediction and molecular dynamics simula-tions. HM and JL performed the major experiments containing site-directed mutagenesis and enzyme production. HM and YM purified and characterized the xylanases. JZ coordinated the study. NH and HZ wrote the manuscript. JD and HZ revised this paper. All authors read and approved the final manuscript.
Author details
1 School of Life Sciences, Yunnan Normal University, Kunming 650500, China. 2 Key Laboratory of Enzyme Engineering, Yunnan Normal University,
Kun-ming 650500, China.
Acknowledgements
We would like to thank Kevin Shi from the Massachusetts Institute of Technol-ogy for providing language help.
Competing interests
The authors declare that they have no competing interests. Additional file
Additional file 1: Figure S1. SDS-PAGE analysis of the recombinant xylanases. Lanes 1, 3, 5 and 7 correspond to purified XynCDBFV, N207A, G208S, and A210S from E. coli BL21 (DE3), respectively; lanes 2, 4, 6 and 8 correspond to expressed XynCDBFV, N207A, G208S, and A210S, respectively; lane 9 corresponds to control cell (harboring empty pEasy-E2 vector); lane M corresponds to standard protein molecular mass markers.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Consent for publication
All authors consent for publication.
Funding
This study is supported by Yunling Scholar [2015] No. 56, Yunling Technology Leading Talent [2014] No. 1782, Applied Basic Research Foundation of Yunnan Province No. 2016FD018, and National Natural Science Foundation of China No. 31660304.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in pub-lished maps and institutional affiliations.
Received: 21 February 2017 Accepted: 17 May 2017
References
1. Scheller HV, Ulvskov P. Hemicelluloses. Annu Rev Plant Biol. 2010;61:263–89.
2. Collins T, Gerday C, Feller G. Xylanases, xylanase families and extremo-philic xylanases. FEMS Microbiol Rev. 2005;29:3–23.
3. Khandeparker R, Numan MT. Bifunctional xylanases and their potential use in biotechnology. J Ind Microbiol Biotechnol. 2008;35:635–44. 4. Taibi Z, Saoudi B, Boudelaa M, Trigui H, Belghith H, Gargouri A, Ladjama
A. Purification and biochemical characterization of a highly thermosta-ble xylanase from Actinomadura sp strain Cpt20 isolated from poultry compost. Appl Biochem Biotech. 2012;166:663–79.
5. Nie SX, Wang SF, Qin CR, Yao SQ, Ebonka JF, Song XP, Li KC. Removal of hexenuronic acid by xylanase to reduce adsorbable organic halides for-mation in chlorine dioxide bleaching of bagasse pulp. Bioresour Technol. 2015;196:413–7.
6. Haki GD, Rakshit SK. Developments in industrially important thermosta-ble enzymes: a review. Bioresour Technol. 2003;89:17–34.
7. Viikari L, Alapuranen M, Puranen T, Vehmaanpera J, Siika-Aho M. Thermo-stable enzymes in lignocellulose hydrolysis. Adv Biochem Eng Biotechnol. 2007;108:121–45.
8. Chen CC, Ko TP, Huang JW, Guo RT. Heat- and alkaline-stable xylanases: application, protein structure and engineering. ChemBioEng Rev. 2015;2:95–106.
9. Berrin JG, Juge N. Factors affecting xylanase functionality in the degrada-tion of arabinoxylans. Biotechnol Lett. 2008;30:1139–50.
10. Polizeli MLTM, Rizzatti ACS, Monti R, Terenzi HF, Jorge JA, Amorim DS. Xylanases from fungi: properties and industrial applications. Appl Micro-biol Biotechnol. 2005;67:577–91.
11. Dumon C, Varvak A, Wall MA, Flint JE, Lewis RJ, Lakey JH, Morland C, Lug-inbuhl P, Healey S, Todaro T, et al. Engineering hyperthermostability into a GH11 xylanase is mediated by subtle changes to protein structure. J Biol Chem. 2008;283:22557–64.
12. Lombard V, Ramulu HG, Drula E, Coutinho PM, Henrissat B. The
carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014;42:D490–5.
13. Paes G, Berrin JG, Beaugrand J. GH11 xylanases: structure/function/prop-erties relationships and applications. Biotechnol Adv. 2012;30:564–92. 14. Purmonen M, Valjakka J, Takkinen K, Laitinen T, Rouvinen J. Molecular dynamics studies on the thermostability of family 11 xylanases. Protein Eng Des Sel. 2007;20:551–9.
• We accept pre-submission inquiries
• Our selector tool helps you to find the most relevant journal • We provide round the clock customer support
• Convenient online submission • Thorough peer review
• Inclusion in PubMed and all major indexing services • Maximum visibility for your research
Submit your manuscript at www.biomedcentral.com/submit
Submit your next manuscript to BioMed Central
and we will help you at every step:
16. Joo JC, Pack SP, Kim YH, Yoo YJ. Thermostabilization of Bacillus circulans xylanase: computational optimization of unstable residues based on thermal fluctuation analysis. J Biotechnol. 2011;151:56–65. 17. Yin X, Li JF, Wang JQ, Tang CD, Wu MC. Enhanced thermostability of a
mesophilic xylanase by N-terminal replacement designed by molecular dynamics simulation. J Sci Food Agric. 2013;93:3016–23.
18. Gao SJ, Wang JQ, Wu MC, Zhang HM, Yin X, Li JF. Engineering hyper-thermostability into a mesophilic family 11 xylanase from Aspergillus oryzae by in silico design of N-terminus substitution. Biotechnol Bioeng. 2013;110:1028–38.
19. Zhang HM, Li JF, Wang JQ, Yang YJ, Wu MC. Determinants for the improved thermostability of a mesophilic family 11 xylanase predicted by computational methods. Biotechnol Biofuels. 2014;7:3.
20. Cheng YS, Chen CC, Huang CH, Ko TP, Luo WH, Huang JW, Liu JR, Guo RT. Structural analysis of a glycoside hydrolase family 11 xylanase from Neocallimastix patriciarum. J Biol Chem. 2014;289:11020–8.
21. Hakulinen N, Turunen O, Janis J, Leisola M, Rouvinen J. Three-dimensional structures of thermophilic beta-1,4-xylanases from Chaetomium thermo-philum and Nonomuraea flexuosa—comparison of twelve xylanases in relation to their thermal stability. Eur J Biochem. 2003;270:1399–412. 22. Ringe D, Petsko GA. Study of protein dynamics by X-ray diffraction.
Meth-ods Enzymol. 1986;131:389–433.
23. Parthasarathy S, Murthy MR. Protein thermal stability: insights from atomic displacement parameters (B values). Protein Eng. 2000;13:9–13. 24. Gruber K, Klintschar G, Hayn M, Schlacher A, Steiner W, Kratky C.
Ther-mophilic xylanase from Thermomyces lanuginosus: high-resolution X-ray structure and modeling studies. Biochemistry. 1998;37:13475–85. 25. Jaenicke R, Schurig H, Beaucamp N, Ostendorp R. Structure and stability
of hyperstable proteins: glycolytic enzymes from hyperthermophilic bacterium Thermotoga maritima. Adv Protein Chem. 1996;48:181–269. 26. Richardson JS, Richardson DC. Natural beta-sheet proteins use negative
design to avoid edge-to-edge aggregation. Proc Natl Acad Sci USA. 2002;99:2754–9.
27. Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–637.
28. Rogers TA, Bommarius AS. Utilizing simple biochemical measurements to predict lifetime output of biocatalysts in continuous isothermal pro-cesses. Chem Eng Sci. 2010;65:2118–24.
29. Podar M, Reysenbach AL. New opportunities revealed by biotechnologi-cal explorations of extremophiles. Curr Opin Biotechnol. 2006;17:250–5. 30. Reetz MT, Carballeira JD, Vogel A. Iterative saturation mutagenesis on
the basis of B factors as a strategy for increasing protein thermostability. Angew Chem Int Ed Engl. 2006;45:7745–51.
31. Kumar PR, Eswaramoorthy S, Vithayathil PJ, Viswamitra MA. The tertiary structure at 1.59 A resolution and the proposed amino acid sequence of a family-11 xylanase from the thermophilic fungus Paecilomyces varioti bainier. J Mol Biol. 2000;295:581–93.
32. Harris GW, Pickersgill RW, Connerton I, Debeire P, Touzel JP, Breton C, Perez S. Structural basis of the properties of an industrially relevant thermo-philic xylanase. Proteins. 1997;29:77–86.
33. Chen YL, Tang TY, Cheng KJ. Directed evolution to produce an alkalophilic variant from a Neocallimastix patriciarum xylanase. Can J Microbiol. 2001;47:1088–94.
34. Guo RT, Chou LJ, Chen YC, Chen CY, Pari K, Jen CJ, Lo SJ, Huang SL, Lee CY, Chang TW, Chaung WJ. Expression in Pichia pastoris and charac-terization by circular dichroism and NMR of rhodostomin. Proteins. 2001;43:499–508.
35. Huang Z, Liu X, Zhang S, Liu Z. GH52 xylosidase from Geobacillus stearothermophilus: characterization and introduction of xylanase activ-ity by site-directed mutagenesis of Tyr509. J Ind Microbiol Biotechnol. 2014;41:65–74.
36. Miller GL. Use of dinitrosalicylic acid reagent for determination of reduc-ing sugar. Anal Chem. 1959;31:426–8.
37. Lineweaver H, Burk D. The determination of enzyme dissociation con-stants. J Am Chem Soc. 1934;56:658–66.
38. Tronrud DE. Knowledge-based B-factor restraints for the refinement of proteins. J App Cryst. 1996;29:100–4.
39. Yuan Z, Zhao J, Wang ZX. Flexibility analysis of enzyme active sites by crystallographic temperature factors. Protein Eng. 2003;16:109–14. 40. Parthasarathy S, Murthy MRN. Analysis of temperature factor distribution
in high-resolution protein structures. Protein Sci. 1997;6:2561–7. 41. Carugo O, Argos P. Accessibility to internal cavities and ligand binding
sites monitored by protein crystallographic thermal factors. Proteins Struct Funct Genet. 1998;31:201–13.
42. Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201.
43. Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Com-parison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–35.
44. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–12.
45. Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. Com-parison of multiple amber force fields and development of improved protein backbone parameters. Proteins. 2006;65:712–25.
46. Hess B, Kutzner C, van der Spoel D, Lindahl E. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theory Comput. 2008;4:435–47.
47. Hess B, Bekker H, Berendsen HJC, Fraaije JGEM. LINCS: a linear constraint solver for molecular simulations. J Comput Chem. 1997;18:1463–72. 48. Darden T, York D, Pedersen L. Particle Mesh Ewald: an N. log(N) method