Genomic Analysis of Multidrug-Resistant
Escherichia coli
from North Carolina
Community Hospitals: Ongoing
Circulation of CTX-M-Producing
ST131-H
30Rx and ST131-
H
30R1 Strains
Hajime Kanamori,
a,bChristian M. Parobek,
cJonathan J. Juliano,
aJames R. Johnson,
d,eBrian D. Johnston,
d,eTimothy J. Johnson,
fDavid J. Weber,
a,bWilliam A. Rutala,
a,bDeverick J. Anderson
g,hDivision of Infectious Diseases, University of North Carolina School of Medicine, Chapel Hill, North Carolina, USAa; Department of Hospital Epidemiology, University of North Carolina Health Care, Chapel Hill, North Carolina, USAb; Curriculum in Genetics and Molecular Biology, University of North Carolina School of Medicine, Chapel Hill, North Carolina, USAc; Division of Infectious Diseases, University of Minnesota, Minneapolis, Minnesota, USAd; Minneapolis Veterans Affairs Health Care System, Minneapolis, Minnesota, USAe; Department of Veterinary and Biomedical Sciences, University of Minnesota, St. Paul, Minnesota, USAf; Division of Infectious Diseases, Duke University Medical Center, Durham, North Carolina, USAg; Duke Center for Antimicrobial Stewardship and Infection Prevention, Durham, North Carolina, USAh
ABSTRACT
Escherichia coli
sequence type 131 (ST131) predominates globally among
multidrug-resistant (MDR)
E. coli
strains. We used whole-genome sequencing (WGS)
to investigate 63 MDR
E. coli
isolates from 7 North Carolina community hospitals
(2010 to 2015). Of these, 39 (62%) represented ST131, including 37 (95%) from the
ST131-
H
30R subclone: 10 (27%) from its
H
30R1 subset and 27 (69%) from its
H
30Rx
subset. ST131 core genomes differed by a median of 15 (range, 0 to 490)
single-nucleotide variants (SNVs) overall versus only 7 within
H
30R1 (range, 3 to 12 SNVs)
and 11 within
H
30Rx (range, 0 to 21). The four isolates with identical core genomes
were all
H
30Rx. Epidemiological and clinical characteristics did not vary significantly
by strain type, but many patients with MDR
E. coli
or
H
30Rx infection were critically
ill and had poor outcomes.
H
30Rx isolates characteristically exhibited
fluoroquin-olone resistance and CTX-M-15 production, had a high prevalence of
trimethoprim-sulfamethoxazole resistance (89%),
sul1
(89%), and
dfrA17
(85%), and were enriched
for specific virulence traits, and all qualified as extraintestinal pathogenic
E. coli
. The
high overall prevalence of CTX-M-15 appeared to be possibly attributable to its
asso-ciation with the ST131-
H
30Rx subclone and IncF[F2:A1:B
⫺
] plasmids. Some
phyloge-netically clustered non-ST131 MDR
E. coli
isolates also had distinctive serotypes/
fimH
types, fluoroquinolone mutations, CTX-M variants, and IncF types. Thus, WGS analysis
of our community hospital source MDR
E. coli
isolates suggested ongoing circulation
and differentiation of
E. coli
ST131 subclones, with clonal segregation of CTX-M
vari-ants, other resistance genes, Inc-type plasmids, and virulence genes.
KEYWORDS
extended-spectrum

-lactamase (ESBL), CTX-M, multidrug-resistant
(MDR)
Escherichia coli
, whole-genome sequencing, community hospitals
M
ultidrug-resistant (MDR)
Escherichia coli
, including strains that produce
extended-spectrum

-lactamases (ESBLs), are a growing clinical and public health
chal-lenge. Since 2000, CTX-M enzymes have become the most prevalent ESBL type in
Gram-negative species, particularly among
E. coli
and other
Enterobacteriaceae
(1).
Currently,
E. coli
sequence type (ST) 131 (ST131), associated with CTX-M-15-type
ESBLs, predominates among MDR
E. coli
strains in the United States and worldwide
Received1 May 2017 Returned for
modification20 May 2017 Accepted1 June
2017
Accepted manuscript posted online5 June
2017
CitationKanamori H, Parobek CM, Juliano JJ,
Johnson JR, Johnston BD, Johnson TJ, Weber DJ, Rutala WA, Anderson DJ. 2017. Genomic analysis of multidrug-resistantEscherichia coli from North Carolina community hospitals: ongoing circulation of CTX-M-producing ST131-H30Rx and ST131-H30R1 strains. Antimicrob Agents Chemother 61:e00912-17.
https://doi.org/10.1128/AAC.00912-17.
Copyright© 2017 American Society for
Microbiology.All Rights Reserved.
Address correspondence to Hajime Kanamori, [email protected].
(1–3). Its
H
30 subclone underlies the ST131 expansion (2). We showed previously that
CTX-M-producing
E. coli
ST131 was prevalent among ESBL-producing isolates from
community hospitals (4). Few other reports address ESBL-producing
E. coli
from U.S.
community settings (5, 6).
Whole-genome sequencing (WGS), an emerging tool for advanced molecular
epidemi-ological investigations involving MDR organisms, allows identification of extended
resis-tance and virulence genotypes and elucidation of transmission routes (7–13). Recently,
WGS-based phylogenetic analyses of
E. coli
ST131 strains identified a CTX-M-15-associated
sublineage, designated
H
30Rx, within ST131-
H
30R, which is the
fluoroquinolone-resistant subset within ST131-
H
30 (11). Such findings suggest that
H
30Rx contributes
importantly to extensive antimicrobial resistance.
Here, we used WGS to investigate the clinical and molecular epidemiology of recent
MDR
E. coli
isolates, especially CTX-M-producing
E. coli
ST131, from patients with
clinically significant infections in North Carolina community hospitals. We also analyzed
resistance genes, virulence genes, and plasmid types in relation to phylogeny.
(This work was presented in part at IDWeek2016, New Orleans, LA [poster
presen-tation no. 200].)
RESULTS
WGS of the 63
E. coli
study isolates generated an average of 3,560,352 read pairs per
isolate. Metagenomic sequence classification confirmed all the isolates as
E. coli
. After
reference-guided assembly, all samples had
ⱖ
10
⫻
coverage over
ⱖ
75% of the
ge-nome, and 62/63 (98%) had
ⱖ
25
⫻
coverage over
ⱖ
75% of the genome (see Fig. S1 in
the supplemental material). Therefore, nearly all the genome assemblies were suitable
for single-nucleotide variant (SNV) calling.
ST distribution.
According to
in silico
multilocus sequence typing (MLST), ST131
was the most prevalent ST, accounting for 39 (62%) of the 63 study isolates. Among the
remaining 24 isolates (38%), the most prevalent ST was ST405 (6; 25% of the non-ST131
isolates; 10% overall). The
fimH
30 allele was present in 36 (92%) of the 39 ST131 isolates
versus only 2 (8%) of the 24 non-ST131 isolates (
P
⬍
0.001). By phylogroup, whereas all
the ST131 isolates were from group B2 (by definition), the non-ST131 isolates were
from, in descending frequency, groups D (
n
⫽
10: 2 ST38, 2 ST69, and 6 ST405), A (
n
⫽
5: 1 ST44, 1 ST167, 2 ST1284, and 1 ST1294), F (
n
⫽
3: 3 ST648), B2 (
n
⫽
3: 2 ST12 and
1 ST1193), B1 (
n
⫽
2: 2 ST224), and undefined (
n
⫽
1: 1 ST3057). By serotype, the 39
ST131 isolates were either O25:H4 (
n
⫽
38 [97%]: 36
fimH
30 and 2
fimH
35) or O16:H5
(
n
⫽
1 [3%]:
fimH
41), whereas the non-ST131 isolates exhibited other serotypes,
including the 6 ST405 isolates (all O102:H6).
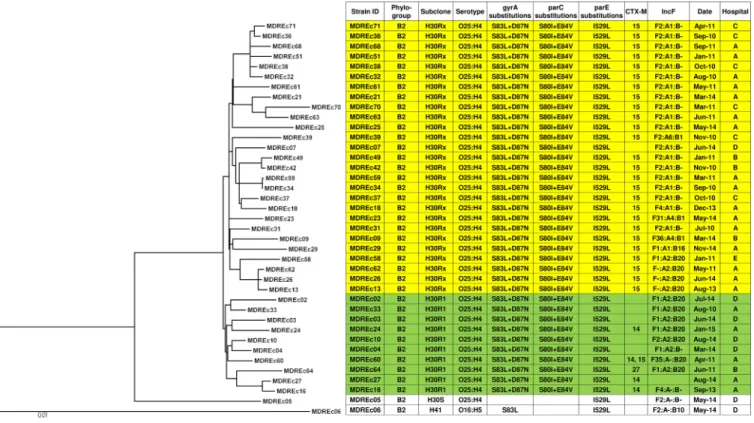
SNV-based phylogeny.
In the core genome phylogeny, the
H
30R1 and
H
30Rx
clades were placed within a larger
H
30R clade (Fig. 1), and other STs (i.e., ST12, ST38,
ST69, ST224, ST405, ST648, and ST1284) demonstrated similar sub-ST differentiation
(Fig. 2). Of the 39 ST131 isolates, 37 (95%) were
H
30R; 10 (27%) of these were
H
30R1 and
27 (73%) were
H
30Rx, including 2 (MDREc63 and MDREc70) that were classified as
H
30Rx despite having
fimH
35, since both were placed with the other
H
30Rx isolates
in the WGS phylogeny. In pairwise comparisons, the median number of SNV
differences between different core genomes was 15 within ST131 overall (range, 0
to 490), including only 7 within
H
30R1 (range, 3 to 12 SNVs) and 11 within
H
30Rx
(range, 0 to 21), compared with 6,627 (range, 12 to 15,128) for the non-ST131 genomes
overall and 24 (range, 13 to 36) for the ST405 genomes.
same clone from our community, or at different times (MDREc13, MDREc26, and
MDREc62; MDREc34 and MDREc59), suggesting local endemicity for specific clones.
Patient characteristics.
Of the 63 source patients for the study isolates (Table 1), 37
(59%) were admitted from home and 29 (46%) had a urinary tract infection. Community
FIG 1Maximum-likelihood phylogram with metadata for 39 multidrug-resistantE. coliST131 strains. The tree with the highest log likelihood (⫺14,477.1472) is shown. The tree is drawn to scale, with branch lengths measured in numbers of substitutions per site. The analysis involved 39 nucleotide sequences. All positions containing gaps and missing data were eliminated. The final data set contained 4,015 total positions. Yellow shading,H30Rx isolates; green shading,H30R1 isolates.
onset health care-associated infections were the most common (30 [48%]), followed by
community-acquired infections (22 [35%]); hospital-acquired infections were
uncom-mon (6 [10%]). Forty-nine (78%) patients survived, while 5 (8%) died during
hospital-ization. Clinical and epidemiological characteristics did not vary significantly in relation
to strain type, except for an association of non-
H
30Rx ST131 isolates with hospital D.
TABLE 1Clinical and epidemiological characteristics of 63 community hospital patients with multidrug-resistantE. coliinfections
Categorya Specific variableb
No. of isolates (column %)
Total (nⴝ63)
ST131 (nⴝ39)
Non-ST131 (nⴝ24)
ST131 H30Rx (nⴝ27)
ST131 non-H30Rx (nⴝ12)
All non-H30Rx (nⴝ36)
Age⬎65 38 (60) 24 (62) 14 (58) 16 (59) 8 (67) 22 (61)
Sex Male 30 (48) 19 (49) 11 (46) 14 (52) 5 (42) 16 (44) Female 26 (41) 18 (46) 8 (33) 11 (41) 7 (58) 15 (42) Unknown 7 (11) 2 (5) 5 (21) 2 (7) 0 5 (14)
Race African American 19 (30) 12 (31) 7 (29) 8 (30) 4 (33) 11 (31) American Indian 1 (2) 0 1 (4) 0 0 1 (3)
Asian 1 (2) 0 1 (4) 0 0 1 (3)
Caucasian 33 (52) 23 (59) 10 (42) 15 (56) 8 (67) 18 (50)
Hispanic 1 (2) 1 (3) 0 1 (4) 0 0
Unknown 8 (13) 3 (8) 5 (21) 3 (11) 0 5 (14)
Community hospital A 35 (56) 21 (54) 14 (58) 16 (59) 5 (42) 19 (53) B 6 (10) 4 (10) 2 (8) 3 (11) 1 (8) 3 (8) C 10 (16) 6 (15) 4 (17) 6 (22) 0 4 (11) D 8 (13) 7 (18) 1 (4) 1 (4) 6 (50) 7 (19)
E 1 (2) 1 (3) 0 1 (4) 0 0
F 1 (2) 0 1 (4) 0 0 1 (3)
G 2 (3) 0 2 (8) 0 0 2 (6)
Admission source Home 37 (59) 21 (54) 16 (67) 12 (44) 9 (75) 25 (69) Nursing home 13 (21) 10 (26) 3 (13) 8 (30) 2 (17) 5 (14) Transfer from hospital 4 (6) 3 (8) 1 (4) 2 (7) 1 (8) 2 (6) Other extended-care facility 3 (5) 3 (8) 1 (4) 3 (11) 0 1 (3) Unknown 5 (8) 2 (5) 3 (13) 2 (7) 0 3 (8)
Type of infection Urinary tract infection 29 (46) 19 (49) 10 (42) 13 (48) 6 (50) 16 (44) Bloodstream infection 21 (33) 14 (36) 7 (29) 9 (33) 5 (42) 12 (33)
Pneumonia 1 (2) 0 1 (4) 0 0 1 (3)
Other 7 (11) 4 (10) 3 (13) 3 (11) 1 (8) 4 (11) Unknown 5 (8) 2 (5) 3 (13) 2 (7) 0 3 (8)
Acquisition Community acquired 22 (35) 14 (36) 8 (33) 8 (30) 6 (50) 14 (39) Community onset, health care associated 30 (48) 20 (51) 10 (42) 15 (56) 5 (42) 15 (42) Hospital onset, health care associated 6 (10) 3 (8) 3 (13) 2 (7) 1 (8) 4 (11) Unknown 5 (8) 2 (5) 3 (13) 2 (7) 0 3 (8)
ICU stay Before infection 4 (6) 2 (5) 2 (8) 1 (4) 1 (8) 3 (8) After infection 46 (73) 30 (77) 16 (67) 20 (74) 10 (83) 26 (72) Unknown 13 (21) 7 (18) 6 (25) 6 (22) 1 (8) 7 (19)
Dialysis Yes 4 (6) 3 (8) 1 (4) 3 (11) 0 1 (3) No 51 (81) 32 (82) 19 (79) 20 (74) 12 (100) 31 (86) Unknown 8 (13) 4 (10) 4 (17) 4 (15) 0 4 (11)
Outcome Died 5 (8) 4 (10) 1 (4) 3 (11) 1 (8) 2 (6) Survived 49 (78) 30 (77) 19 (79) 20 (74) 10 (83) 29 (81) Discharged home 22 (35) 15 (38) 7 (29) 9 (33) 6 (50) 13 (36) Discharged to hospice 5 (8) 2 (5) 3 (13) 2 (7) 0 3 (8) Discharged to LTCF 16 (25) 11 (28) 5 (21) 8 (30) 3 (25) 8 (22) Discharged to other facility 6 (10) 2 (5) 4 (17) 1 (4) 1 (8) 5 (14) Unknown 9 (14) 5 (13) 4 (17) 4 (15) 1 (8) 5 (14)
aCategory of clinical or epidemiological characteristic.
Resistance phenotypes and genotypes.
Although most or all isolates were
sus-ceptible to imipenem, ertapenem, amikacin, and minocycline, most (80 to 90%) were
resistant to trimethoprim-sulfamethoxazole and fluoroquinolones (ciprofloxacin and
levofloxacin) (Table 2 and Fig. 3), drugs that are commonly used orally to treat urinary
tract infections in community hospitals (4). Compared to non-
H
30Rx ST131 isolates, or
all non-
H
30Rx isolates,
H
30Rx isolates had higher resistance prevalences for most
antibiotics (Table 2). Accordingly, they had significantly higher aggregate resistance
scores (median score [interquartile range], 6 [5 to 6] for
H
30Rx versus 3.5 [3 to 4] for
non-
H
30Rx ST131 isolates [
P
⬍
0.001] and 4 [3 to 6] for all non-
H
30Rx isolates [
P
⫽
0.02]).
WGS analysis identified multiple resistance genes and plasmids, plus associations of
bla
CTX-Mwith diverse classes of antimicrobial resistance (Fig. 3; see Table S1 in the
supplemental material). Whereas CTX-M-15 was closely associated with
H
30Rx isolates
(
P
⬍
0.001), CTX-M-14 was associated with non-
H
30Rx ST131 isolates, both overall (
P
⫽
0.002) and within ST131 (
P
⫽
0.006) (Table 2). All 37
H
30R isolates contained
charac-teristic
H
30-associated quinolone resistance-determining region (QRDR) mutations,
including
gyrA
1A1B1 (S83L/D87N),
parC
1a1A1B1 (S80I/E84V), and
parE
3L (I529L) (
P
⬍
0.001 for
H
30Rx versus all non-
H
30Rx isolates and for ST131 versus non-ST131).
Additionally, 15 (56%) of the 27
H
30Rx isolates, but only 1 (8%) of the 12 non-
H
30Rx
ST131 isolates, possessed the plasmid-mediated quinolone resistance determinant
aac(6
=
)-Ib-cr
(
P
⫽
0.01).
H
30Rx isolates also had a significantly higher frequency than did
all non-
H
30Rx isolates of certain non-quinolone resistance genes (i.e.,
aadA1
,
aadA5
,
catB3
, and
dfrA17
).
Virulence genes.
ST131 (versus non-ST131) and
H
30Rx (versus non-
H
30Rx) isolates
were significantly enriched with specific virulence genes (i.e.,
pap
,
sfa
,
yfcV
,
iss
,
kps
,
iha
,
iuc
,
iutA
,
sat
, and
malX
) (Table 3; see Table S2 in the supplemental material).
Accord-ingly, they had significantly higher aggregate virulence scores (median score
[inter-quartile range], 22 [21 to 23] for ST131 versus 14 [12.25 to 17.5] for non-ST131,
P
⬍
0.001, and 22 [21 to 23] for
H
30Rx versus 15.5 [13 to 21] for all non-
H
30Rx,
P
⬍
0.001).
Likewise, a much greater proportion qualified as extraintestinal pathogenic
E. coli
(ExPEC), i.e., 37 (95%) of 39 ST131 isolates, including all 27
H
30Rx isolates, versus 8
(33%) of 24 non-ST131 isolates (
P
⬍
0.001 for all comparisons). However, despite their
differences in virulence gene content, these strain groups did not differ significantly in
associated clinical outcomes (Table 1). Regardless of the strain type, 46 (73%) of 63
patients were admitted to an intensive care unit (ICU) after infection, and 10 (16%) died
or were discharged to hospice.
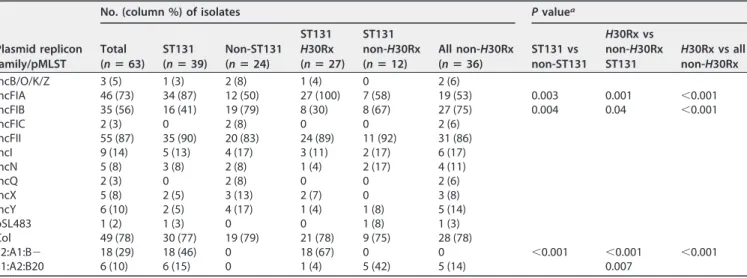
Plasmid replicon families/types.
H
30Rx isolates contained IncFIA plasmid types
more frequently, and IncFIB plasmid types less frequently, than other isolates (
P
⬍
0.001) (Table 4).
In silico
pMLST associated IncF[F2:A1:B
⫺
] with
H
30Rx and IncF[F1:A2:
B20] with
H
30R1, whereas neither of these IncF types appeared in non-ST131 isolates.
Other results for non-ST131 isolates.
The non-ST131 isolates exhibited
associa-tions analogous to those noted among the ST131 isolates between specific STs and
distinctive serotypes/
fimH
types (for ST12, ST38, ST69, ST224, ST405, ST648, and
ST1284), QRDR mutations (for ST12, ST224, ST405, ST648, and ST1284), CTX-M variants
(for ST1284, ST224, ST405, and ST648), and IncF types (for ST224 and ST648) (Fig. 2). All
ST405 isolates had identical QRDR mutations (
gyrA
20A1B1 [S83L/D87N],
parC
5A1
[S80I], and
parE
1A1 [S458A]), which were distinct from those associated with ST131.
DISCUSSION
We used WGS to analyze MDR
E. coli
isolates collected over 6 years from community
hospitals in North Carolina to identify molecular markers of resistance, virulence, and
transmission. To our knowledge, this is the first WGS study of clinical MDR
E. coli
isolates
from community hospital inpatients.
TABLE 2 Antimicrobial resistance characteristics among 63 multidrug-resistant E. coli isolates from community hospitals Antimicrobial category Resistance gene/antimicrobial agent No. (column %) of isolates P value c
Total (n
ⴝ
63)
ST131 (n
ⴝ
39)
Non-ST131 (n
ⴝ
24)
ST131 H30Rx (n
and the typically more extensive resistance profiles of
H
30Rx isolates compared with
other ST131 isolates, including even members of the
fluoroquinolone-resistance-associated
H
30R1 subclone (11).
We also documented clonal segregation of CTX-M variants, other resistance genes,
and Inc-type plasmids, both within ST131 and among non-ST131 isolates. With finer
resolution than MLST, WGS-based genotyping identified a variety of SNVs among
isolates from the same ST or even sub-ST clade, demonstrating mainly dissemination of
diverse
H
30R1/
H
30Rx strains throughout the study community rather than focal
nos-ocomial or community outbreaks affecting a single community hospital. The
endemic-ity documented here suggested that
H
30R1/
H
30Rx strains were circulating prior to the
study period. Based on previous epidemiological data, the introduction of these strains
likely occurred from 2006 through 2007, when the rate of ESBL-producing
E. coli
infection in our network of community hospitals rose from 0 to 12 patients per 100,000
patient-days (14).
The SNV-based phylogeny showed differentiation of our ST131 strains into the
previously described
H
30R1 and
H
30Rx clades (11–13). Corresponding to this clonal
structure, we identified the IncFIA and IncFII (e.g., IncF[F2:A1:B
⫺
]) replicons in most
H
30Rx isolates that produced CTX-M-15. While ST131 can harbor ESBL-encoding
plas-mids of different incompatibility groups,
bla
CTX-M-15has been associated primarily with
IncF, especially FIA-FII fusion or FII replicons (3). A recent WGS study of ST131 isolates
from the United States likewise described associations of IncF[F2:A1:B
⫺
] with
H
30Rx
and IncF[F1:A2:B20] with
H
30R1, demonstrating fixation and ongoing evolution of
specific plasmids within these separate but closely related sublineages (15). Our data
extend these findings to community hospital source ST131 isolates.
All
H
30R study isolates had distinctive
H
30R-associated double nonsynonymous
mutations in
gyrA
and
parC
(16–18), which confer high-level quinolone resistance
(13). The
H
30Rx strains additionally harbored
aac(6
=
)Ib-cr
(encoding a dual-function
aminoglycoside- and fluoroquinolone-modifying enzyme) significantly more frequently
than other strains, supporting an association of the gene specifically with
H
30Rx (18).
They also exhibited a high prevalence of trimethoprim-sulfamethoxazole resistance
(89%),
sul1
(89%), and
dfrA17
(85%) and had higher aggregate resistance scores than
other strains, consistent with other reports from the United States and elsewhere (9, 19,
TABLE 3Putative virulence genes among 63 multidrug-resistantE. coliisolates from community hospitals
Category
Virulence gene
No. (column %) of isolates Pvaluea
Total (nⴝ63)
ST131 (nⴝ39)
Non-ST131 (nⴝ24)
ST131 H30Rx (nⴝ27)
ST131 non-H30Rx (nⴝ12)
All non-H30Rx (nⴝ36)
ST131 vs non-ST131
H30Rx vs all non-H30Rx
Adhesin dra 13 (21) 12 (31) 1 (4) 8 (30) 4 (33) 5 (14) 0.01 fim 59 (94) 39 (100) 20 (83) 27 (100) 12 (100) 32 (89) 0.02 nfaE 13 (21) 12 (31) 1 (4) 8 (30) 4 (33) 5 (14) 0.01
pap 44 (70) 37 (95) 7 (29) 27 (100) 10 (83) 17 (47) ⬍0.001 ⬍0.001 sfa 44 (70) 37 (95) 7 (29) 27 (100) 10 (83) 17 (47) ⬍0.001 ⬍0.001 yfcV 45 (71) 39 (100) 6 (25) 27 (100) 12 (100) 18 (50) ⬍0.001 ⬍0.001
Protectin iss 48 (76) 38 (97) 10 (42) 27 (100) 11 (92) 21 (58) ⬍0.001 ⬍0.001 kfiC 12 (19) 11 (28) 1 (4) 5 (19) 6 (50) 7 (19) 0.02
kps 47 (75) 38 (97) 9 (38) 27 (100) 11 (92) 20 (56) ⬍0.001 ⬍0.001
Siderophore chu 55 (87) 38 (97) 17 (71) 26 (96) 12 (100) 29 (81) 0.004 fyuA 58 (92) 39 (100) 19 (79) 27 (100) 12 (100) 31 (86) 0.006
iha 39 (62) 36 (92) 3 (13) 26 (96) 10 (83) 13 (36) ⬍0.001 ⬍0.001 irp 58 (92) 39 (100) 19 (79) 27 (100) 12 (100) 31 (86) 0.006
iuc 47 (75) 35 (90) 12 (50) 25 (93) 10 (83) 22 (61) ⬍0.001 0.007 iutA 47 (75) 35 (90) 12 (50) 25 (93) 10 (83) 22 (61) ⬍0.001 0.007
Toxin sat 38 (60) 35 (90) 3 (13) 25 (93) 10 (83) 13 (36) ⬍0.001 ⬍0.001 Miscellaneous malX 53 (84) 39 (100) 14 (58) 27 (100) 12 (100) 26 (72) ⬍0.001 0.003 ExPEC NAb 45 (71) 37 (95) 8 (33) 27 (100) 10 (83) 18 (50) ⬍0.001 ⬍0.001
aOnlyPvalues of⬍0.05 are shown. bNA, not applicable.
TABLE 4Plasmid replicon families and plasmid MLST (pMLST) among 63 multidrug-resistantE. coliisolates from community hospitals
Plasmid replicon family/pMLST
No. (column %) of isolates Pvaluea
Total (nⴝ63)
ST131 (nⴝ39)
Non-ST131 (nⴝ24)
ST131 H30Rx (nⴝ27)
ST131 non-H30Rx (nⴝ12)
All non-H30Rx (nⴝ36)
ST131 vs non-ST131
H30Rx vs non-H30Rx ST131
H30Rx vs all non-H30Rx
IncB/O/K/Z 3 (5) 1 (3) 2 (8) 1 (4) 0 2 (6)
IncFIA 46 (73) 34 (87) 12 (50) 27 (100) 7 (58) 19 (53) 0.003 0.001 ⬍0.001 IncFIB 35 (56) 16 (41) 19 (79) 8 (30) 8 (67) 27 (75) 0.004 0.04 ⬍0.001 IncFIC 2 (3) 0 2 (8) 0 0 2 (6)
IncFII 55 (87) 35 (90) 20 (83) 24 (89) 11 (92) 31 (86) IncI 9 (14) 5 (13) 4 (17) 3 (11) 2 (17) 6 (17) IncN 5 (8) 3 (8) 2 (8) 1 (4) 2 (17) 4 (11)
IncQ 2 (3) 0 2 (8) 0 0 2 (6)
IncX 5 (8) 2 (5) 3 (13) 2 (7) 0 3 (8) IncY 6 (10) 2 (5) 4 (17) 1 (4) 1 (8) 5 (14) pSL483 1 (2) 1 (3) 0 0 1 (8) 1 (3) Col 49 (78) 30 (77) 19 (79) 21 (78) 9 (75) 28 (78)
F2:A1:B⫺ 18 (29) 18 (46) 0 18 (67) 0 0 ⬍0.001 ⬍0.001 ⬍0.001 F1:A2:B20 6 (10) 6 (15) 0 1 (4) 5 (42) 5 (14) 0.007
20). These features may give
H
30Rx strains a competitive advantage over other
ESBL-producing strains, especially with use of fluoroquinolones and extended-spectrum
cephalosporins (21, 22), thereby contributing to their epidemiological success.
As observed previously (23, 24), most ST131 study isolates and all
H
30Rx isolates
qualified as ExPEC, with an extensive array of putative virulence genes and higher
virulence scores, suggesting high virulence potential. However, higher virulence
scores were not associated with worse clinical outcomes. Among ST131/
H
30Rx
strains, host characteristics may confound associations of virulence genes with
clinical severity and/or mortality (21). Nonetheless, in a recent analysis, the
H
30
subclone was significantly associated with persistent infections and adverse
out-comes despite multivariable adjustment for underlying host factors and resistance
to the prescribed antibiotic (21). In our study, a majority of patients with
H
30Rx
were critically ill and had poor outcomes (i.e., 74% frequency of ICU admission after
infection and 11% all-cause mortality), although this did not differ significantly from
other strain groups.
It remains unclear whether the most extensively resistant
E. coli
ST131 strains are
from the community or health care setting (2). In a recent study of unselected
E. coli
clinical isolates from the U.S. Midwest, ST131 was associated with young children and
the elderly, antimicrobial resistance, and health care-associated infections (25). In
contrast, in our North Carolina community hospital-based study of MDR
E. coli
isolates,
we observed no significant difference in patient age or site of infection acquisition
between ST131 and non-ST131 strains or between
H
30Rx and non-
H
30Rx ST131 strains.
Our results establish that health care personnel in community hospitals may encounter
MDR
E. coli
ST131 in patients with community-acquired infection, community onset
health care-associated infections, or hospital onset health care-associated infections.
This indicates a need to facilitate appropriate antimicrobial therapy and infection
control measures against MDR
E. coli
ST131 in community hospitals.
In that regard, approximately 80% of the present community hospital MDR
E. coli
infections were present on admission with or without prior health care exposure,
similar to our previous finding (4). Additionally, various movements of patients with
MDR
E. coli
ST131-
H
30Rx occurred among different health care facilities, both before
admission to and after discharge from our community hospitals (e.g., admission from
home to community hospital and then discharge to home, nursing home, or hospice)
(see Table S3 in the supplemental material). This emphasizes potential routes for strain
dissemination and the important role of community hospitals and
transferring/receiv-ing health care facilities as reservoirs of MDR
E. coli
.
Although previous WGS studies have investigated
E. coli
ST131 (11–13), to our
knowledge, none has addressed the molecular epidemiology of non-ST131 MDR
E. coli
.
Here, although most non-ST131 isolates, like the ST131 isolates, produced CTX-M
variants (e.g., CTX-M-15 or CTX-M-14) and exhibited quinolone resistance, they had
different QRDR mutations and IncF types than did the ST131 strains. Although ST131 is
currently the most prevalent MDR
E. coli
strain in our community and globally, attention
to resistance in non-ST131
E. coli
also is clearly warranted.
This study has several limitations. First, limited clinical and epidemiological data
were available from our surveillance databases, although data entry and definitions
were standardized across participating hospitals. Second, our sample was derived from
multiple centers over a 6-year period, adding breadth to our analysis, but at the
expense of depth. While this reduced our power to detect meaningful associations, it
may increase the generalizability of our findings. Third, our small sample size limited
the power to confirm statistical associations, particularly for non-ST131 isolates.
dom-inance of
H
30Rx strains carrying IncF[F2:A1:B
⫺
] plasmids suggests that this lineage
represents an especially successful combination of extensive resistance and virulence.
Further genomic-epidemiological studies of
E. coli
ST131 and its
H
30R1 and
H
30Rx
subclones are needed in community hospitals to inform antimicrobial stewardship
programs, empirical therapy approaches, and infection control strategies rooted in the
community.
MATERIALS AND METHODS
Bacterial isolates.The study sites were 7 geographically dispersed community hospitals in or near North Carolina affiliated with the Duke Infection Control Outreach Network (DICON). Between 2010 and 2015, local infection preventionists prospectively used Centers for Disease Control and Prevention surveillance definitions (26) to identify all hospitalized patients with MDRE. coliinfections. Isolates were classified as MDR if they were nonsusceptible toⱖ1 agent inⱖ3 antimicrobial classes (27). The hospital laboratories confirmed ESBL production phenotypically by using Clinical and Laboratory Standards Institute (CLSI) criteria. Duke University’s Institutional Review Board approved the study.
During 2010 to 2015, the study hospitals reported 100 total MDR E. coliinfections. Sixty-three nonduplicate MDRE. coliisolates (54 were ESBL producers), including 61 from 6 North Carolina hospitals and 2 from 1 Virginia hospital, were saved in the DICON Biorepository and subsequently sent to the University of North Carolina at Chapel Hill (UNC) for further analysis.
Clinical and epidemiological data.Clinical and epidemiological data for the source patients were obtained through the DICON database. Variables included age, sex, race, admission source, type of infection, setting for infection acquisition, presence/absence of ICU stay, dialysis use, and discharge outcome.
Antimicrobial susceptibility testing.The UNC Clinical Laboratories reassessed isolates’ susceptibil-ity to 12 antibiotics by disk diffusion according to the 2015 CLSI guideline (28). Isolates classified as intermediate or resistant were regarded as resistant.
Whole-genome sequencing.Isolates were grown overnight in LB broth at 37°C. Total DNA was extracted and purified using an UltraClean microbial DNA isolation kit (Mo Bio, Carlsbad, CA). Sequencing libraries were prepared using the NEBNext Ultra DNA Library Prep kit for Illumina (New England BioLabs, Ipswich, MA) with NEBNext Multiplex oligonucleotides (New England BioLabs). Size-fractionated DNA libraries were labeled, pooled, and sequenced at the UNC High-Throughput Sequencing Facility in a single Illumina HiSeq2500 run using 125-base paired-end V4 chemistry.
Sequence assembly and mapping.De novoand reference-guided assembly methods were used for gene identification and comparative genetic analysis, respectively, unless otherwise noted. De novo assemblies were compiled using the A5-miseq pipeline with default parameter settings (29). Reference-guided assemblies againstE. colistrain EC958 (GenBank accession no.HG941718.1) were compiled using bwa mem(30) (see Fig. S1 in the supplemental material). Assemblies were deduplicated and subjected to local realignment through high-entropic regions using Picard (http://broadinstitute.github.io/picard/) and the Genome Analysis Toolkit (GATK) v3.3 (31).
Variant calling.SNVs were identified by applying the GATK UnifiedGenotyper across all reference-aligned isolates simultaneously. The SNVs were filtered stringently using cutoffs responsive to the underlying distribution of quality scores. For filtering, the parameters included the following: quality by depth,ⱖ25; mapping quality,ⱖ59; Fisher score,ⱕ4; map quality rank sum,ⱖ⫺4.0; and read position rank sum,ⱖ⫺4.0.
Genetic analysis. Species identification was confirmed by k-mer-based classification based on short-read sequences using Kraken (32). Phylogenetic groups were inferred from known ST assignments (33). Specific genes and alleles thereof were identified via the Center for Genomic Epidemiology’s bioinformatic pipeline (http://www.genomicepidemiology.org/), using default settings except where otherwise stated. Specifically, serotypeFinder 1.1 (34) was used for serotypes, FimTyper 1.0 (35) forfimH types, MLST v1.7 (36) and the Achtman MLST scheme (http://mlst.warwick.ac.uk/mlst/dbs/Ecoli) (37) for in silico MLST, ResFinder v2.1 (38) for acquired resistance genes (threshold, 98% identity; minimum length, 60%), PlasmidFinder 1.3 for plasmid incompatibility groups (threshold, 95% identity), and pMLST 1.4 (39) for plasmid STs. The resistance score was the number of antimicrobial classes for which an isolate was resistant toⱖ1 representative agent (40).
ForE. coliST131 isolates, subclonesH30R (ciprofloxacin resistant;fimHallele 30),H30Rx (a CTX-M-15-associated clonal subset withinH30R), andH30R1 (a sister clade toH30Rx withinH30R) were classified as described previously (11, 15).H30Rx was defined based on a specific SNV,ybbWG723A, as identified by BLASTn (http://blast.ncbi.nlm.nih.gov/Blast.cgi) (19).
To investigate QRDRs, established alleles ofgyrA,parC, andparEwere identified using agyrA,parC, parEallele database generously provided by E. Sokurenko, University of Washington. Stepwise muta-tional derivatives of each numbered allele ingyrA,parC, andparEwere identified.
For phylogenetic analysis, recombinant genomic regions were identified using Gubbins (43) and were excluded. A maximum-likelihood (ML) tree was inferred using the Tamura-Nei model (44) within MEGA7 (45) (use of the general time-reversible substitution model yielded a similar tree). The ML phylogeny was constructed from genetic variation in nonrecombinant genomic regions. The initial tree(s) for the heuristic search was obtained automatically by applying the neighbor-joining and BioNJ algorithms to a matrix of pairwise distances estimated using the maximum composite likelihood (MCL) approach and then selecting the topology with superior log likelihood value. Pairwise SNV differences between core genomes were calculated using bedtools (https://bedtools .readthedocs.io/en/latest/).
Statistical analysis.Using JMP 11 (Statistical Analysis System, Cary, NC, USA), categorical variables were compared by the two-tailed Fisher test or the chi-square test, and continuous variables were evaluated by the Wilcoxon test. The significance threshold was aPvalue of⬍0.05.
Accession number(s).Sequence data have been deposited in the DDBJ/EMBL/GenBank Sequence Read Archive (SRA) under accession numbersDRX055674toDRX055737.
SUPPLEMENTAL MATERIAL
Supplemental material for this article may be found at
https://doi.org/10.1128/AAC
.00912-17
.
SUPPLEMENTAL FILE 1,
PDF file, 0.4 MB.
ACKNOWLEDGMENTS
We are grateful to Veronika L. Tchesnokova and Evgeni V. Sokurenko for their
assistance with assignment of
gyrA
,
parC
, and
parE
alleles. We also acknowledge Nicole
Stoesser for providing her list of virulence sequences.
The opinions expressed here are strictly ours and do not necessarily reflect those of
our respective institutions or the Department of Veterans Affairs.
H.K. received financial support from a Japan Society for the Promotion of Science
(JSPS) Postdoctoral Fellowship for Research Abroad. C.M.P. was supported by NIH
training grant AI109979. This material is also based in part on work supported by Office
of Research and Development, Medical Research Service, Department of Veterans
Affairs grant 1 I01 CX000920-01 (J.R.J.). D.J.A. received support from the NIH/NIAID
(K23AI095357).
J.R.J. has ST131-related research grants from Actavis/Allergan, Merck, and
Tetra-phase; a consultancy with Janssen/Crucell; and patent applications for tests related to
specific
E. coli
strains. We report no other conflicts of interest relevant to this article.
REFERENCES
1. Mathers AJ, Peirano G, Pitout JD. 2015. The role of epidemic resistance plasmids and international high-risk clones in the spread of multidrug-resistantEnterobacteriaceae. Clin Microbiol Rev 28:565–591.https://doi .org/10.1128/CMR.00116-14.
2. Banerjee R, Johnson JR. 2014. A new clone sweeps clean: the enigmatic emergence ofEscherichia coli sequence type 131. Antimicrob Agents Chemother 58:4997–5004.https://doi.org/10.1128/AAC.02824-14. 3. Nicolas-Chanoine MH, Bertrand X, Madec JY. 2014.Escherichia coliST131,
an intriguing clonal group. Clin Microbiol Rev 27:543–574.https://doi .org/10.1128/CMR.00125-13.
4. Chen LF, Freeman JT, Nicholson B, Keiger A, Lancaster S, Joyce M, Woods CW, Cook E, Adcock L, Louis S, Cromer AL, Sexton DJ, Anderson DJ. 2014. Widespread dissemination of CTX-M-15 genotype extended-spectrum- -lactamase-producingEnterobacteriaceae among patients presenting to community hospitals in the southeastern United States. Antimicrob Agents Chemother 58:1200 –1202.https://doi.org/10.1128/AAC.01099-13. 5. Banerjee R, Strahilevitz J, Johnson JR, Nagwekar PP, Schora DM, Shevrin I,
Du H, Peterson LR, Robicsek A. 2013. Predictors and molecular epidemiol-ogy of community-onset extended-spectrum-lactamase-producing Esch-erichia coliinfection in a Midwestern community. Infect Control Hosp Epidemiol 34:947–953.https://doi.org/10.1086/671725.
6. Doi Y, Park YS, Rivera JI, Adams-Haduch JM, Hingwe A, Sordillo EM, Lewis JS, II, Howard WJ, Johnson LE, Polsky B, Jorgensen JH, Richter SS, Shutt KA, Paterson DL. 2013. Community-associated extended-spectrum
-lactamase-producing Escherichia coliinfection in the United States. Clin Infect Dis 56:641– 648.https://doi.org/10.1093/cid/cis942. 7. Kanamori H, Parobek CM, Weber DJ, van Duin D, Rutala WA, Cairns BA,
Juliano JJ. 2015. Next-generation sequencing and comparative analysis
of sequential outbreaks caused by multidrug-resistantAcinetobacter bau-manniiat a large academic burn center. Antimicrob Agents Chemother 60:1249 –1257.https://doi.org/10.1128/AAC.02014-15.
8. Salipante SJ, SenGupta DJ, Cummings LA, Land TA, Hoogestraat DR, Cookson BT. 2015. Application of whole-genome sequencing for bacterial strain typing in molecular epidemiology. J Clin Microbiol 53:1072–1079.https://doi.org/10.1128/JCM.03385-14.
9. Sherchan JB, Hayakawa K, Miyoshi-Akiyama T, Ohmagari N, Kirikae T, Na-gamatsu M, Tojo M, Ohara H, Sherchand JB, Tandukar S. 2015. Clinical epidemiology and molecular analysis of extended-spectrum- -lactamase-producingEscherichia coliin Nepal: characteristics of sequence types 131 and 648. Antimicrob Agents Chemother 59:3424 –3432.https://doi.org/10 .1128/AAC.00270-15.
10. Stoesser N, Sheppard AE, Moore CE, Golubchik T, Parry CM, Nget P, Saroeun M, Day NP, Giess A, Johnson JR, Peto TE, Crook DW, Walker AS, Modernizing Medical Microbiology Informatics Group. 2015. Ex-tensive within-host diversity in fecally carried extended-spectrum- -lactamase-producingEscherichia coliisolates: implications for trans-mission analyses. J Clin Microbiol 53:2122–2131.https://doi.org/10 .1128/JCM.00378-15.
11. Price LB, Johnson JR, Aziz M, Clabots C, Johnston B, Tchesnokova V, Nordstrom L, Billig M, Chattopadhyay S, Stegger M, Andersen PS, Pear-son T, Riddell K, Rogers P, Scholes D, Kahl B, Keim P, Sokurenko EV. 2013. The epidemic of extended-spectrum--lactamase-producingEscherichia coliST131 is driven by a single highly pathogenic subclone,H30-Rx. mBio 4:e00377-13.https://doi.org/10.1128/mBio.00377-13.
Dougan G, Rodriguez-Bano J, Pascual A, Pitout JD, Upton M, Paterson DL, Walsh TR, Schembri MA, Beatson SA. 2014. Global dissemination of a multidrug resistantEscherichia coliclone. Proc Natl Acad Sci U S A 111:5694 –5699.https://doi.org/10.1073/pnas.1322678111.
13. Stoesser N, Sheppard AE, Pankhurst L, De Maio N, Moore CE, Sebra R, Turner P, Anson LW, Kasarskis A, Batty EM, Kos V, Wilson DJ, Phetsou-vanh R, Wyllie D, Sokurenko E, Manges AR, Johnson TJ, Price LB, Peto TE, Johnson JR, Didelot X, Walker AS, Crook DW. 2016. Evolutionary history of the global emergence of the Escherichia coli epidemic clone ST131. mBio 7:e02162-15.https://doi.org/10.1128/mBio.02162-15.
14. Freeman JT, Sexton DJ, Anderson DJ. 2009. Emergence of extended-spectrum-lactamase-producingEscherichia coliin community hospitals throughout North Carolina: a harbinger of a wider problem in the United States? Clin Infect Dis 49:e30 – e32.https://doi.org/10.1086/600046. 15. Johnson TJ, Danzeisen JL, Youmans B, Case K, Llop K, Munoz-Aguayo J,
Flores-Figueroa C, Aziz M, Stoesser N, Sokurenko E, Price LB, Johnson JR, Castanheira M. 2016. Separate F-type plasmids have shaped the evolu-tion of the H30 subclone ofEscherichia colisequence type 131. mSphere 1:e00121-16.https://doi.org/10.1128/mSphere.00121-16.
16. Colpan A, Johnston B, Porter S, Clabots C, Anway R, Thao L, Kuskowski MA, Tchesnokova V, Sokurenko EV, Johnson JR, VICTORY (Veterans Influence of Clonal Types on Resistance: Year 2011) Investigators. 2013. Escherichia colisequence type 131 (ST131) subcloneH30 as an emergent multidrug-resistant pathogen among US veterans. Clin Infect Dis 57: 1256 –1265.https://doi.org/10.1093/cid/cit503.
17. Johnson JR, Tchesnokova V, Johnston B, Clabots C, Roberts PL, Billig M, Riddell K, Rogers P, Qin X, Butler-Wu S, Price LB, Aziz M, Nicolas-Chanoine MH, Debroy C, Robicsek A, Hansen G, Urban C, Platell J, Trott DJ, Zhanel G, Weissman SJ, Cookson BT, Fang FC, Limaye AP, Scholes D, Chattopadhyay S, Hooper DC, Sokurenko EV. 2013. Abrupt emergence of a single dominant multidrug-resistant strain ofEscherichia coli. J Infect Dis 207:919 –928.https://doi.org/10.1093/infdis/jis933.
18. Peirano G, van der Bij AK, Freeman JL, Poirel L, Nordmann P, Costello M, Tchesnokova VL, Pitout JD. 2014. Characteristics ofEscherichia coli se-quence type 131 isolates that produce extended-spectrum-lactamases: global distribution of theH30-Rx sublineage. Antimicrob Agents Che-mother 58:3762–3767.https://doi.org/10.1128/AAC.02428-14. 19. Banerjee R, Robicsek A, Kuskowski MA, Porter S, Johnston BD, Sokurenko
E, Tchesnokova V, Price LB, Johnson JR. 2013. Molecular epidemiology of Escherichia colisequence type 131 and ItsH30 andH30-Rx subclones among extended-spectrum--lactamase-positive and -negative E. coli clinical isolates from the Chicago region, 2007 to 2010. Antimicrob Agents Chemother 57:6385– 6388.https://doi.org/10.1128/AAC.01604-13. 20. Matsumura Y, Johnson JR, Yamamoto M, Nagao M, Tanaka M, Takakura
S, Ichiyama S, Kyoto-Shiga Clinical Microbiology Study Group. 2015. CTX-M-27- and CTX-M-14-producing, ciprofloxacin-resistantEscherichia coliof theH30 subclonal group within ST131 drive a Japanese regional ESBL epidemic. J Antimicrob Chemother 70:1639 –1649.https://doi.org/ 10.1093/jac/dkv017.
21. Johnson JR, Thuras P, Johnston BD, Weissman SJ, Limaye AP, Riddell K, Scholes D, Tchesnokova V, Sokurenko E. 2016. The pandemicH30 sub-clone ofEscherichia colisequence type 131 is associated with persistent infections and adverse outcomes independent from its multidrug resis-tance and associations with compromised hosts. Clin Infect Dis 62: 1529 –1536.https://doi.org/10.1093/cid/ciw193.
22. Banerjee R, Johnston B, Lohse C, Porter SB, Clabots C, Johnson JR. 2013. Escherichia colisequence type 131 is a dominant, antimicrobial-resistant clonal group associated with healthcare and elderly hosts. Infect Control Hosp Epidemiol 34:361–369.https://doi.org/10.1086/669865.
23. Johnson JR, Menard M, Johnston B, Kuskowski MA, Nichol K, Zhanel GG. 2009. Epidemic clonal groups ofEscherichia coli as a cause of anti-microbial-resistant urinary tract infections in Canada, 2002 to 2004. Antimicrob Agents Chemother 53:2733–2739.https://doi.org/10.1128/ AAC.00297-09.
24. Olesen B, Frimodt-Moller J, Leihof RF, Struve C, Johnston B, Hansen DS, Scheutz F, Krogfelt KA, Kuskowski MA, Clabots C, Johnson JR. 2014. Temporal trends in antimicrobial resistance and virulence-associated traits within theEscherichia colisequence type 131 clonal group and its H30 andH30-Rx subclones, 1968 to 2012. Antimicrob Agents Chemother 58:6886 – 6895.https://doi.org/10.1128/AAC.03679-14.
25. Banerjee R, Johnston B, Lohse C, Chattopadhyay S, Tchesnokova V, Sokurenko EV, Johnson JR. 2013. The clonal distribution and diversity of extraintestinalEscherichia coliisolates vary according to patient
charac-teristics. Antimicrob Agents Chemother 57:5912–5917.https://doi.org/ 10.1128/AAC.01065-13.
26. Centers for Disease Control and Prevention, National Healthcare Safety Network. January 2017. CDC/NHSN surveillance definitions for specific types of infections.https://www.cdc.gov/nhsn/pdfs/pscmanual/17pscnosinfdef _current.pdf. Accessed 24 April 2017.
27. Magiorakos AP, Srinivasan A, Carey RB, Carmeli Y, Falagas ME, Giske CG, Harbarth S, Hindler JF, Kahlmeter G, Olsson-Liljequist B, Paterson DL, Rice LB, Stelling J, Struelens MJ, Vatopoulos A, Weber JT, Monnet DL. 2012. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: an international expert proposal for interim standard definitions for acquired resistance. Clin Microbiol Infect 18:268 –281.
https://doi.org/10.1111/j.1469-0691.2011.03570.x.
28. Clinical and Laboratory Standards Institute. 2015. Performance standards for antimicrobial susceptibility testing; 20th informational supplement M100-S25. Clinical and Laboratory Standards Institute, Wayne, PA. 29. Coil DJG, Darling AE. 2015. A5-miseq: an updated pipeline to
assem-ble microbial genomes from Illumina MiSeq data. Bioinformatics 31: 587–589.https://doi.org/10.1093/bioinformatics/btu661.
30. Li H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv:1303.3997 [q-bio.GN].https://arxiv.org/ abs/1303.3997.
31. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20:1297–1303.https:// doi.org/10.1101/gr.107524.110.
32. Wood DE, Salzberg SL. 2014. Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol 15:R46.https://doi .org/10.1186/gb-2014-15-3-r46.
33. Clermont O, Gordon D, Denamur E. 2015. Guide to the various phylo-genetic classification schemes forEscherichia coliand the correspon-dence among schemes. Microbiology 161:980 –988.https://doi.org/10 .1099/mic.0.000063.
34. Joensen KG, Tetzschner AM, Iguchi A, Aarestrup FM, Scheutz F. 2015. Rapid and easyin silicoserotyping ofEscherichia coliisolates by use of whole-genome sequencing data. J Clin Microbiol 53:2410 –2426.https:// doi.org/10.1128/JCM.00008-15.
35. Sokurenko E, Tchesnokova V, Muradova M, Chattophadhay S, Ahrenfeldt J, Allesøe R, Hansen F, Hammerum A, Hasman H. 2016. Fimtyper; a Web tool forE. coli fimHgenotyping, abstr PO8. Abstr 11th Int Meet Microb Epidemiol Markers (IMMEM XI), Estoril, Portugal.https://www.escmid .org/fileadmin/src/media/PDFs/3Research_Projects/Conferences/IMMEM _11_Abstracts_Book.pdf.
36. Larsen MV, Cosentino S, Rasmussen S, Friis C, Hasman H, Marvig RL, Jelsbak L, Sicheritz-Ponten T, Ussery DW, Aarestrup FM, Lund O. 2012. Multilocus sequence typing of total-genome-sequenced bacteria. J Clin Microbiol 50:1355–1361.https://doi.org/10.1128/JCM.06094-11. 37. Wirth T, Falush D, Lan R, Colles F, Mensa P, Wieler LH, Karch H, Reeves PR,
Maiden MC, Ochman H, Achtman M. 2006. Sex and virulence in Esche-richia coli: an evolutionary perspective. Mol Microbiol 60:1136 –1151.
https://doi.org/10.1111/j.1365-2958.2006.05172.x.
38. Zankari E, Hasman H, Cosentino S, Vestergaard M, Rasmussen S, Lund O, Aarestrup FM, Larsen MV. 2012. Identification of acquired antimicrobial resistance genes. J Antimicrob Chemother 67:2640 –2644. https://doi .org/10.1093/jac/dks261.
39. Carattoli A, Zankari E, García-Fernández A, Voldby Larsen M, Lund O, Villa L, Møller Aarestrup F, Hasman H. 2014.In silicodetection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob Agents Chemother 58:3895–3903.https://doi.org/10.1128/ AAC.02412-14.
40. Johnson JR, Urban C, Weissman SJ, Jorgensen JH, Lewis JS II, Hansen G, Edelstein PH, Robicsek A, Cleary T, Adachi J, Paterson D, Quinn J, Hanson ND, Johnston BD, Clabots C, Kuskowski MA, AMERECUS Investigators. 2012. Molecular epidemiological analysis ofEscherichia colisequence type ST131 (O25:H4) and blaCTX-M-15 among extended-spectrum-
-lactamase-producingE. colifrom the United States, 2000 to 2009. Antimicrob Agents Chemother 56:2364 –2370.https://doi.org/10.1128/AAC.05824-11. 41. Johnson JR, Russo TA. 2005. Molecular epidemiology of extraintestinal
pathogenic (uropathogenic)Escherichia coli. Int J Med Microbiol 295: 383– 404.https://doi.org/10.1016/j.ijmm.2005.07.005.
chicken products. Antimicrob Agents Chemother 47:2161–2168.https:// doi.org/10.1128/AAC.47.7.2161-2168.2003.
43. Croucher NJ, Page AJ, Connor TR, Delaney AJ, Keane JA, Bentley SD, Parkhill J, Harris SR. 2015. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res 43:e15.https://doi.org/10.1093/nar/gku1196.
44. Tamura K, Nei M. 1993. Estimation of the number of nucleotide substi-tutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol 10:512–526.