organic papers
o2256
Ingebrigtsenet al. C14H10N2 doi:10.1107/S1600536805019306 Acta Cryst.(2005). E61, o2256–o2257
Acta Crystallographica Section E Structure Reports Online
ISSN 1600-5368
4-(2-Naphthyl)pyrimidine
Truls Ingebrigtsen, Tore Lejon and Lars Kr. Hansen*
Department of Chemistry, University of Tromsø, 9037 Tromsø, Norway
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study
T= 298 K
Mean(C–C) = 0.005 A˚
Rfactor = 0.040
wRfactor = 0.097 Data-to-parameter ratio = 9.8
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2005 International Union of Crystallography
Printed in Great Britain – all rights reserved
The title compound, C14H10N2, has been synthesized from the
appropriate ketones and formamide using different palladium complexes as catalysts. The pyrimidine group is twisted 30.48 (9) relative to the naphthalene part of the molecule,
resulting in an intramolecular C—H N hydrogen bond. There is also an intermolecular C—H N hydrogen bond linking the molecules in the crystal structure.
Comment
Pyrimidines are widely found in nature,e.g.in pyrimidine and purine bases in nucleic acids, and in the vitamin thiamin. They have also attracted interest as potential drugs, and are avail-able in the nucleoside analogue AZTTMused in AIDS therapy, AcyclovirTMused in treatment of herpes infections and in the
prodrug CapecitabineTM used in cancer therapy. The
inter-esting chemical and physiological properties of pyrimidines have led to a number of syntheses being developed (von Angerer et al., 2004). In our procedure, good yields of the expected products are formed from the appropriate ketone when reacted with formamide, catalysed by different palla-dium complexes (Ingebrigtsen et al., 2005).
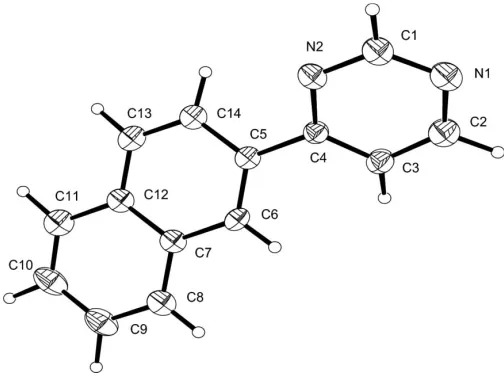
The atomic numbering scheme of the title compound, (I), is shown in Fig. 1. Bond lengths are within the normal range of such bonds (Allen et al., 1987). The least-squares plane through the pyrimidine part of the molecule forms a dihedral angle of 30.48 (9)with the naphthalene residue. This
config-uration allows for a short intramolecular hydrogen bond (C14—H14 N2). There is also a short intermolecular hydrogen bond (C1—H1 N1) contributing to the packing of the molecules in the crystal structure (Taylor & Kennard, 1982). Table 1 lists selected hydrogen bonds shorter than the van der Waals distance (Bondi, 1964).
Experimental
To a 10 ml flask charged with Pd(OAc)2(0.05 equivalents) and PPh3 (0.10 equivalents) were added formamide (5.0 g), PhI (2.0 g) and ketone (1.0 equivalents). The resulting mixture was heated at 433 K for 8 h. The reaction mixture was diluted with diethyl ether and extracted three times with 2MHCl. The combined aqueous layers
were basified with 4MNaOH and extracted with diethyl ether. The organic layer was washed with water and brine and dried over Na2CO3. Evaporation of the solvent gave the crude product as a white solid. Purification by silica column chromatography (EtOAc), gave crystals that were dissolved in a small amount of diethyl ether. Heptane was added and crystals of the title compound were grown by slow evaporation of the solvent at room temperature.
Crystal data
C14H10N2 Mr= 206.24
Monoclinic,P21=a a= 7.4467 (16) A˚
b= 6.1343 (11) A˚
c= 22.720 (3) A˚
= 92.508 (19) V= 1036.9 (3) A˚3
Z= 4
Dx= 1.321 Mg m 3
MoKradiation Cell parameters from 25
reflections
= 12–18 = 0.08 mm1
T= 298 (2) K Plate, white
0.500.300.10 mm
Data collection
Enraf–Nonius CAD-4 diffractometer
!–2scans
Absorption correction: scan [Northet al., (1968) and
ABSCALCinOSCAIL
(McArdle & Daly, 1999)]
Tmin= 0.961,Tmax= 0.992
1984 measured reflections 1813 independent reflections
586 reflections withI> 2(I)
Rint= 0.008 max= 24.9 h=2!8
k= 0!7
l=26!26 3 standard reflections
frequency: 120 min intensity decay: 2%
Refinement
Refinement onF2 R[F2> 2(F2)] = 0.040 wR(F2) = 0.097
S= 0.82 1813 reflections 185 parameters
All H-atom parameters refined
w= 1/[2(F
o2) + (0.0341P)2]
whereP= (Fo2+ 2Fc2)/3 (/)max= 0.035
max= 0.16 e A˚ 3
min=0.17 e A˚ 3
Table 1
Hydrogen-bond geometry (A˚ ,).
D—H A D—H H A D A D—H A
C14—H14 N2 1.01 (2) 2.49 (3) 2.827 (4) 99 (2) C1—H1 N1i 0.93 (2) 2.60 (3) 3.440 (5) 151 (2) C13—H13 C6ii
1.00 (2) 2.78 (3) 3.735 (5) 163 (2) C8—H8 C11iii
0.98 (2) 2.82 (3) 3.716 (3) 153 (2)
Symmetry codes: (i)x1 2;y
1
2;z; (ii)xþ 1 2;yþ
1 2;z; (iii)x
1 2;yþ
3 2;z.
All the H atoms were found in a difference map and were refined independently; C—H = 0.93 (3)–1.07 (3) A˚ . The quality of the crystal was rather poor and accordingly data were collected only tomax= 24.9.
Data collection:CAD-4-PC Software (Enraf–Nonius, 1992); cell refinement: CELDIM in CAD-4-PC Software; data reduction:
XCAD4 (McArdle & Higgins, 1995); program(s) used to solve structure:SHELXS97(Sheldrick, 1997); program(s) used to refine structure: SHELXL97 (Sheldrick, 1997); molecular graphics:
ORTEX (McArdle, 1995); software used to prepare material for publication:OSCAIL(McArdle, 1993).
References
Allen, F. H., Kennard, O., Watson, D. G., Brammer, L., Orpen, A. G. & Taylor, R. (1987).J. Chem. Soc. Perkin Trans. 2, pp. S1–19.
Angerer, S. von (2004).Methods Synth.16, 379–572. Bondi, A. (1964).J. Chem. Phys.68, 441–451.
Enraf–Nonius (1992).CAD-4-PC Software. Version 1.1. Enraf–Nonius, Delft, The Netherlands.
Ingebrigtsen, T., Helland, I. & Lejon, T. (2005).Heterocycles.Submitted. McArdle, P. (1993).J. Appl. Cryst.26, 752.
McArdle, P. (1995).J. Appl. Cryst.28, 65.
McArdle, P. & Daly, P. (1999).ABSCALC.PC version. National University of Ireland, Galway, Ireland.
McArdle, P. & Higgins, T. (1995).XCAD4. National University of Ireland, Galway, Ireland.
North, A. C. T., Phillips, D. C. & Mathews, F. S. (1968).Acta Cryst.A24, 351– 359.
Sheldrick, G. M. (1997). SHELXL97 and SHELXS97. University of Go¨ttingen, Germany.
[image:2.610.313.565.69.261.2]Taylor, R. & Kennard, O. (1982).J. Am. Chem. Soc.104, 5063–5070.
Figure 1
supporting information
sup-1
Acta Cryst. (2005). E61, o2256–o2257
supporting information
Acta Cryst. (2005). E61, o2256–o2257 [https://doi.org/10.1107/S1600536805019306]
4-(2-Naphthyl)pyrimidine
Truls Ingebrigtsen, Tore Lejon and Lars Kr. Hansen
4-(2-Naphthyl)pyrimidine
Crystal data
C14H10N2 Mr = 206.24
Monoclinic, P21/a
a = 7.4467 (16) Å
b = 6.1343 (11) Å
c = 22.720 (3) Å
β = 92.508 (19)°
V = 1036.9 (3) Å3
Z = 4
F(000) = 432
Dx = 1.321 Mg m−3
Mo Kα radiation, λ = 0.71069 Å
Cell parameters from 25 reflections
θ = 12–18°
µ = 0.08 mm−1
T = 298 K
Plate, white
0.50 × 0.30 × 0.10 mm
Data collection
Enraf–Nonius CAD-4 diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
ω–2θ scans
Absorption correction: ψ scan
[North et al., 1968) and ABSCALC in OSCAIL
(McArdle & Daly, 1999)] Tmin = 0.961, Tmax = 0.992 1984 measured reflections
1813 independent reflections 586 reflections with I > 2σ(I) Rint = 0.008
θmax = 24.9°, θmin = 1.8°
h = −2→8
k = 0→7
l = −26→26
3 standard reflections every 120 min intensity decay: 2%
Refinement
Refinement on F2
Least-squares matrix: full R[F2 > 2σ(F2)] = 0.040 wR(F2) = 0.097
S = 0.82
1813 reflections 185 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
All H-atom parameters refined w = 1/[σ2(F
o2) + (0.0341P)2] where P = (Fo2 + 2Fc2)/3 (Δ/σ)max = 0.035
Special details
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Least-squares planes (x,y,z in crystal coordinates) and deviations from them (* indicates atom used to define plane)
- 7.0770 (0.0039) x + 0.0143 (0.0099) y + 8.0080 (0.0290) z = 1.3900 (0.0077)
* -0.0009 (0.0023) N2 * 0.0013 (0.0028) C1 * 0.0035 (0.0027) N1 * -0.0083 (0.0029) C2 * 0.0083 (0.0026) C3 * -0.0038 (0.0022) C4 0.0147 (0.0051) C5
Rms deviation of fitted atoms = 0.0053
6.6260 (0.0036) x + 2.7788 (0.0046) y - 2.1426 (0.0130) z = 1.4254 (0.0047)
Angle to previous plane (with approximate e.s.d.) = 30.48 (0.09)
* -0.0283 (0.0025) C5 * -0.0055 (0.0026) C6 * 0.0221 (0.0028) C7 * 0.0145 (0.0028) C8 * -0.0021 (0.0031) C9 * -0.0191 (0.0031) C10 * -0.0098 (0.0028) C11 * 0.0069 (0.0028) C12 * 0.0188 (0.0029) C13 * 0.0027 (0.0026) C14 - 0.1351 (0.0043) C4
Rms deviation of fitted atoms = 0.0155
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used
only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2
are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
N2 −0.0627 (4) 0.4682 (4) 0.11710 (11) 0.0531 (8)
N1 −0.1464 (4) 0.7324 (6) 0.04330 (12) 0.0653 (9)
H1 −0.153 (4) 0.412 (5) 0.0396 (13) 0.063 (11)*
H14 0.162 (4) 0.280 (4) 0.1833 (12) 0.039 (8)*
H6 −0.042 (3) 0.818 (4) 0.2587 (11) 0.043 (10)*
H10 0.201 (4) 0.401 (4) 0.4754 (13) 0.057 (10)*
H13 0.256 (4) 0.139 (5) 0.2784 (12) 0.061 (11)*
H9 0.053 (4) 0.749 (5) 0.4633 (13) 0.073 (12)*
H8 −0.021 (4) 0.872 (5) 0.3682 (12) 0.052 (11)*
H11 0.258 (4) 0.170 (6) 0.3863 (13) 0.075 (12)*
H3 −0.006 (4) 0.963 (5) 0.1641 (11) 0.048 (9)*
H2 −0.127 (4) 1.047 (5) 0.0651 (13) 0.071 (11)*
C1 −0.1235 (6) 0.5307 (7) 0.06378 (16) 0.0641 (12)
C2 −0.1008 (5) 0.8877 (7) 0.08208 (16) 0.0633 (11)
C3 −0.0383 (5) 0.8425 (6) 0.13905 (16) 0.0486 (10)
C4 −0.0192 (4) 0.6288 (6) 0.15519 (13) 0.0388 (8)
C5 0.0458 (4) 0.5591 (5) 0.21478 (14) 0.0375 (8)
C6 0.0145 (4) 0.6798 (6) 0.26364 (14) 0.0391 (9)
C7 0.0683 (4) 0.6056 (6) 0.32077 (14) 0.0394 (9)
C8 0.0328 (4) 0.7266 (7) 0.37187 (15) 0.0499 (10)
C9 0.0800 (5) 0.6501 (8) 0.42668 (16) 0.0624 (13)
C10 0.1652 (5) 0.4463 (8) 0.43285 (17) 0.0663 (13)
C11 0.2023 (5) 0.3240 (7) 0.38488 (17) 0.0539 (11)
C12 0.1535 (4) 0.4017 (6) 0.32712 (14) 0.0428 (9)
C13 0.1897 (5) 0.2801 (6) 0.27600 (15) 0.0475 (10)
supporting information
sup-3
Acta Cryst. (2005). E61, o2256–o2257 Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
N2 0.065 (2) 0.053 (2) 0.0402 (17) −0.0025 (17) −0.0116 (15) −0.0075 (16)
N1 0.085 (2) 0.061 (2) 0.0483 (19) 0.006 (2) −0.0113 (17) 0.004 (2)
C1 0.087 (3) 0.055 (3) 0.049 (3) −0.002 (3) −0.016 (2) −0.009 (2)
C2 0.078 (3) 0.053 (3) 0.059 (3) 0.007 (3) 0.002 (2) 0.013 (2)
C3 0.054 (2) 0.041 (2) 0.051 (2) −0.003 (2) −0.0005 (19) −0.003 (2)
C4 0.040 (2) 0.037 (2) 0.039 (2) 0.0001 (19) −0.0018 (16) −0.0031 (18)
C5 0.0306 (19) 0.040 (2) 0.042 (2) −0.0026 (18) 0.0015 (16) −0.0025 (17)
C6 0.036 (2) 0.030 (2) 0.050 (3) 0.0011 (18) −0.0050 (18) 0.0003 (17)
C7 0.0266 (19) 0.049 (2) 0.043 (2) −0.0011 (19) −0.0013 (16) 0.0007 (18)
C8 0.040 (2) 0.063 (3) 0.047 (3) −0.002 (2) −0.0015 (19) −0.006 (2)
C9 0.045 (2) 0.098 (4) 0.044 (3) 0.002 (3) −0.002 (2) −0.006 (3)
C10 0.051 (3) 0.105 (4) 0.043 (3) −0.005 (3) −0.003 (2) 0.012 (3)
C11 0.038 (2) 0.060 (3) 0.064 (3) −0.006 (2) −0.003 (2) 0.015 (2)
C12 0.031 (2) 0.050 (2) 0.047 (2) −0.0068 (19) −0.0057 (17) 0.0088 (19)
C13 0.040 (2) 0.043 (3) 0.059 (3) 0.004 (2) 0.001 (2) 0.002 (2)
C14 0.036 (2) 0.047 (2) 0.048 (2) −0.003 (2) 0.0067 (18) −0.007 (2)
Geometric parameters (Å, º)
N2—C1 1.331 (4) C7—C12 1.407 (4)
N2—C4 1.341 (3) C7—C8 1.413 (4)
N1—C1 1.330 (4) C8—C9 1.362 (4)
N1—C2 1.331 (4) C8—H8 0.98 (3)
C1—H1 0.93 (3) C9—C10 1.406 (5)
C2—C3 1.384 (5) C9—H9 1.05 (3)
C2—H2 1.07 (3) C10—C11 1.361 (5)
C3—C4 1.367 (4) C10—H10 1.03 (3)
C3—H3 0.96 (3) C11—C12 1.428 (4)
C4—C5 1.481 (4) C11—H11 1.03 (3)
C5—C6 1.363 (4) C12—C13 1.416 (4)
C5—C14 1.416 (4) C13—C14 1.367 (4)
C6—C7 1.417 (4) C13—H13 1.00 (3)
C6—H6 0.95 (3) C14—H14 1.01 (3)
C1—N2—C4 116.0 (3) C8—C7—C6 121.9 (3)
C1—N1—C2 114.1 (3) C9—C8—C7 121.3 (4)
N1—C1—N2 128.3 (4) C9—C8—H8 118.7 (17)
N1—C1—H1 119.9 (19) C7—C8—H8 119.9 (17)
N2—C1—H1 111.8 (19) C8—C9—C10 119.6 (4)
N1—C2—C3 122.8 (4) C8—C9—H9 118.3 (17)
N1—C2—H2 112.3 (17) C10—C9—H9 122.1 (17)
C3—C2—H2 124.8 (17) C11—C10—C9 121.1 (4)
C4—C3—C2 118.0 (4) C11—C10—H10 123.2 (17)
C4—C3—H3 124.1 (17) C9—C10—H10 115.6 (16)
N2—C4—C3 120.8 (3) C10—C11—H11 125.1 (18)
N2—C4—C5 116.0 (3) C12—C11—H11 114.9 (18)
C3—C4—C5 123.2 (3) C7—C12—C13 119.0 (3)
C6—C5—C14 119.0 (3) C7—C12—C11 119.1 (4)
C6—C5—C4 121.8 (3) C13—C12—C11 121.9 (4)
C14—C5—C4 119.1 (3) C14—C13—C12 120.2 (4)
C5—C6—C7 121.3 (3) C14—C13—H13 118.2 (16)
C5—C6—H6 118.7 (16) C12—C13—H13 121.6 (17)
C7—C6—H6 120.0 (16) C13—C14—C5 121.2 (4)
C12—C7—C8 118.9 (3) C13—C14—H14 123.7 (15)
C12—C7—C6 119.2 (3) C5—C14—H14 115.0 (15)
C2—N1—C1—N2 0.3 (6) C12—C7—C8—C9 0.3 (5)
C4—N2—C1—N1 0.1 (6) C6—C7—C8—C9 177.8 (3)
C1—N1—C2—C3 −1.2 (6) C7—C8—C9—C10 −0.3 (5)
N1—C2—C3—C4 1.6 (6) C8—C9—C10—C11 0.4 (6)
C1—N2—C4—C3 0.3 (5) C9—C10—C11—C12 −0.4 (5)
C1—N2—C4—C5 179.1 (3) C8—C7—C12—C13 −179.9 (3)
C2—C3—C4—N2 −1.1 (5) C6—C7—C12—C13 2.6 (4)
C2—C3—C4—C5 −179.8 (3) C8—C7—C12—C11 −0.4 (4)
N2—C4—C5—C6 −148.1 (3) C6—C7—C12—C11 −178.0 (3)
C3—C4—C5—C6 30.7 (5) C10—C11—C12—C7 0.4 (5)
N2—C4—C5—C14 29.5 (4) C10—C11—C12—C13 179.9 (3)
C3—C4—C5—C14 −151.7 (3) C7—C12—C13—C14 −2.1 (5)
C14—C5—C6—C7 −1.3 (4) C11—C12—C13—C14 178.5 (3)
C4—C5—C6—C7 176.4 (3) C12—C13—C14—C5 −0.1 (5)
C5—C6—C7—C12 −0.9 (4) C6—C5—C14—C13 1.8 (5)
C5—C6—C7—C8 −178.4 (3) C4—C5—C14—C13 −175.9 (3)
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
C14—H14···N2 1.01 (2) 2.49 (3) 2.827 (4) 99 (2)
C1—H1···N1i 0.93 (2) 2.60 (3) 3.440 (5) 151 (2)
C13—H13···C6ii 1.00 (2) 2.78 (3) 3.735 (5) 163 (2)
C8—H8···C11iii 0.98 (2) 2.82 (3) 3.716 (3) 153 (2)