metal-organic papers
m1940
Brayet al. C2H8N4O6Pd doi:10.1107/S1600536805027224 Acta Cryst.(2005). E61, m1940–m1942 Acta Crystallographica Section E
Structure Reports
Online
ISSN 1600-5368
(Ethane-1,2-diamine)dinitratopalladium(II)
David J. Bray,a,bJack K. Clegg,b* Li-Ling Liao,c,bLeonard F. Lindoy,b John C. McMurtrie,d,b David Schilter,bGang Weia,band Tae-Jin Wonb
aCSIRO, Industrial Physics, Bradfield Road,
West Lindfield, New South Wales 2070, Australia,bCentre for Heavy Metals Research, School of Chemistry, F11, The University of Sydney, NSW 2006, Australia,cSchool of Physics and Chemistry, Guizhou Normal University, Guiyang, Guizhou, People’s Republic of China, anddSchool of Physical and
Chemical Sciences, Queensland University of Technology, Queensland 4001, Australia
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study
T= 150 K
Mean(C–C) = 0.002 A˚
Rfactor = 0.016
wRfactor = 0.043
Data-to-parameter ratio = 15.0
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2005 International Union of Crystallography Printed in Great Britain – all rights reserved
The title compound, [Pd(NO3)2(C2H8N2)], forms an infinite
two-dimensional sheet-like motif, propagated by inter-molecular hydrogen bonds between the amino groups of the ethane-1,2-diamine ligands and the nitrate O atoms. There are two complex molecules in the asymmetric unit.
Comment
Our group has long been interested in the use of metal complexes as components for the construction of large supramolecular architectures (Lindoy & Atkinson, 2000). In particular, we are interested in the construction and chemistry of metallocyclic systems (Clegg et al., 2004, 2005). The title compound, (I), has found extensive use as a precursor in the preparation of cyclic metallo-supramolecular structures (Fujita et al., 2005). Crystals suitable for this study were obtained in the course of our investigation into the inter-actions of N-donor ligand systems (Brayet al., 2005) with (I).
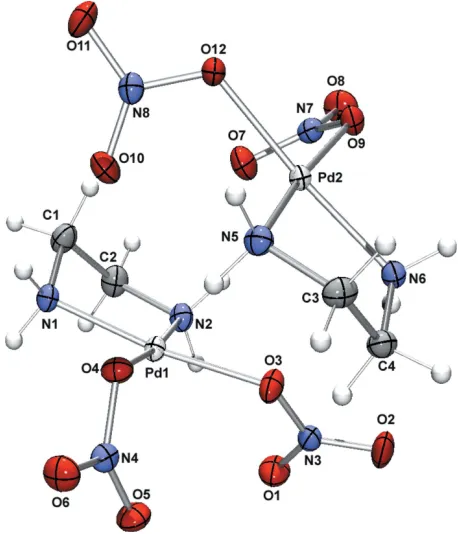
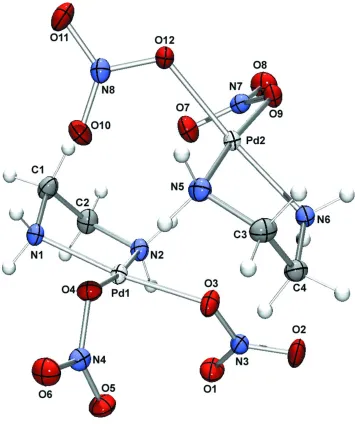
An ORTEP(Farrugia, 1997) representation of (I) is given in Fig. 1. As expected, each PdIIion has a geometry close to an ideal square-plane (Table 1). The N donor atoms of the bidentate ethane-1,2-diamine ligand (en) occupy two coordi-nation sites in a typical five-membered chelate arrangment. The remaining coordination sites are occupied by nitrate O atoms of two nitrate ligands.
The asymmetric unit contains two of these complexes, which pack via intermolecular hydrogen bonds between the NH2
groups of the en ligands and the O atoms of the nitrate ligands. Hydrogen-bond details are provided in Table 2.
The intermolecular hydrogen bonds propagate in two dimensions, forming an infinite sheet-like motif that lies parallel to thebcplane (Fig. 2). Each of the N donor atoms forms hydrogen bonds to (at least) two O acceptor atoms, with only atoms O5 and O8 not involved in close interactions. The sheets stack along theaaxis, as shown in the crystal packing diagram (Fig. 3).
Experimental
The title compound was prepared fromcis-[Pd(en)Cl2] and identified as the desired product by comparison with literature data (Fujitaet al., 1996; Tercero-Morenoet al., 1996). Crystals of (I) suitable for the X-ray diffraction study were isolated from methanol after several days of slow evaporation. All reagents were purchased from Sigma– Aldrich.
Crystal data
C2H8N4O6Pd
Mr= 290.52
Monoclinic,P21=c
a= 16.8478 (6) A˚
b= 7.7746 (3) A˚
c= 13.0702 (5) A˚
= 109.816 (1)
V= 1610.62 (10) A˚3
Z= 8
Dx= 2.396 Mg m
3
MoKradiation
Cell parameters from 10524 reflections
= 2.4–28.3
= 2.32 mm1
T= 150 (2) K Block, colourless 0.510.300.16 mm
Data collection
Bruker SMART 1000 CCD area-detector diffractometer
!scans
Absorption correction: multi-scan (SADABS; Sheldrick, 1999)
3881 independent reflections 3554 reflections withI> 2(I)
Rint= 0.019
max= 28.3
[image:2.610.342.534.73.359.2]h=22!22 Figure 1
[image:2.610.56.285.74.341.2]A representation of the asymmetric unit of (I), shown with 50% probability displacement ellipsoids.
Figure 2
Figure 3
[image:2.610.84.258.388.704.2]Refinement
Refinement onF2 R[F2> 2(F2)] = 0.016
wR(F2) = 0.043
S= 1.03 3881 reflections 259 parameters
H atoms treated by a mixture of independent and constrained refinement
w= 1/[2
(Fo2) + (0.0236P)2 + 0.7251P]
whereP= (Fo2+ 2Fc2)/3 (/)max= 0.002
max= 1.08 e A˚ 3
[image:3.610.43.295.209.308.2]min=0.40 e A˚ 3
Table 1
Selected geometric parameters (A˚ ,).
N1—Pd1 2.0032 (14)
N2—Pd1 2.0231 (14)
N5—Pd2 2.0102 (15)
N6—Pd2 2.0150 (14)
O3—Pd1 2.0326 (12)
O4—Pd1 2.0492 (11)
O9—Pd2 2.0465 (12)
O12—Pd2 2.0426 (11)
N1—Pd1—N2 83.84 (6)
N1—Pd1—O3 172.80 (6)
N2—Pd1—O3 94.62 (5)
N1—Pd1—O4 90.81 (5)
N2—Pd1—O4 173.19 (5)
O3—Pd1—O4 90.16 (5)
N5—Pd2—N6 83.82 (6)
N5—Pd2—O12 95.14 (5)
N6—Pd2—O12 173.68 (5)
N5—Pd2—O9 173.01 (5)
N6—Pd2—O9 91.35 (5)
[image:3.610.44.296.357.499.2]O12—Pd2—O9 89.11 (5)
Table 2
Hydrogen-bond geometry (A˚ ,).
D—H A D—H H A D A D—H A
N2—H3 O7 0.90 (2) 2.23 (2) 2.9167 (19) 134 (2) N2—H3 O11i
0.90 (2) 2.45 (2) 3.0911 (19) 129 (2)
N2—H4 O1 0.89 (2) 2.49 (2) 3.009 (2) 118 (2)
N2—H4 O9ii
0.89 (2) 2.58 (2) 3.415 (2) 158 (2) N5—H6 O4 0.88 (2) 2.45 (2) 3.2837 (19) 160 (2) N5—H6 O4 0.88 (2) 2.45 (2) 3.2837 (19) 160 (2) N1—H2 O11ii
0.86 (2) 2.35 (2) 3.199 (2) 167 (2) N1—H2 O12ii
0.86 (2) 2.46 (2) 3.1177 (19) 133 (2) N1—H1 O9iii
0.88 (2) 2.24 (2) 3.0642 (18) 157 (2) N6—H7 O3 0.88 (2) 2.39 (2) 2.9550 (18) 122 (2) N6—H7 O10i
0.88 (2) 2.50 (2) 3.1324 (19) 130 (2)
N6—H7 O2 0.88 (2) 2.58 (2) 3.363 (2) 149 (2)
N6—H8 O4i 0.88 (2) 2.38 (2) 3.1695 (18) 150 (2) N6—H8 O6i
0.88 (2) 2.40 (2) 3.1628 (19) 145 (2)
Symmetry codes: (i)x;yþ3 2;z
1
2; (ii)x;y1;z; (iii)x;yþ 3 2;zþ
1 2.
C-bound H atoms were included in idealized positions and refined using a riding-model approximation, with methylene C—H bond lengths fixed at 0.99 A˚ . N-bound H atoms were located in a difference Fourier map and refined with bond-length restraints of 0.90 (2) A˚ . Uiso(H) values were fixed at 1.2Ueq(C) and 1.5Ueq(N). The maximum residual electron-density peak is located 0.86 A˚ from atom Pd2.
Data collection:SMART(Bruker, 1995); cell refinement:SAINT (Bruker, 1995); data reduction:SAINTandXPREP(Bruker, 1995); program(s) used to solve structure:SIR97 (Altomare et al., 1999); program(s) used to refine structure:SHELXL97(Sheldrick, 1997); molecular graphics: ORTEP3 (Farrugia, 1997) and WinGX32 (Farrugia, 1999); software used to prepare material for publication: enCIFer(Allenet al., 2004).
The authors gratefully acknowledge the Australian Research Council for financial support.
References
Allen, F. H., Johnson, O., Shields, G. P., Smith, B. R. & Towler, M. (2004).J. Appl. Cryst.37, 335–338.
Altomare, A., Burla, M. C., Camalli, M., Cascarano, G., Giacovazzo, C., Guagliardi, A., Moliterni, A. G. G., Polidori, G. & Spagna, R. (1999).J. Appl. Cryst.32, 115–119.
Bray, D. J., Liao, L.-L., Antonioli, B., Gloe, K., Lindoy, L. F., McMurtrie, J. C., Wei, G. & Zhang, X.-Y. (2005).Dalton Trans.pp. 2082–2083.
Bruker (1995).SMART(Version 5.054),SAINT(Version 6.45) andXPREP
(Version 6.02). Bruker AXS Inc., Madison, Wisconsin, USA.
Clegg, J. K., Lindoy, L. F., McMurtrie, J. C., Moubaraki, B. & Murray, K. (2004).Dalton Trans.pp. 2417–2423.
Clegg, J. K., Lindoy, L. F., McMurtrie, J. C. & Schilter, D. (2005).Dalton Trans.
pp. 857–864.
Farrugia, L. J. (1997).J. Appl. Cryst.30, 565. Farrugia, L. J. (1999).J. Appl. Cryst.32, 837–838.
Fujita, M., Aoyagi, M. & Ogura, K. (1996).Inorg. Chim. Acta,246, 53–57. Fujita, M., Tominaga, M., Hori, A. & Therrien, B. (2005).Acc. Chem. Res.38,
369–378.
Lindoy, L. F. & Atkinson, I. M. (2000).Self-Assembly in Supramolecular Systems. Monographs in Supramolecular Chemistry, Series Editor J. F. Stoddart. Cambridge: Royal Society of Chemistry.
Sheldrick, G. M. (1997).SHELXL97. University of Go¨ttingen, Germany. Sheldrick, G. M. (1999).SADABS. University of Go¨ttingen, Germany. Tercero-Moreno, A. M.-H. J. M., Gorza´lez-Garcia´, S. & Niclo´s-Gutie´rrez, J.
(1996).Inorg. Chim. Acta,253, 23–29.
metal-organic papers
m1942
Brayet al. Csupporting information
Acta Cryst. (2005). E61, m1940–m1942 [doi:10.1107/S1600536805027224]
(Ethane-1,2-diamine)dinitratopalladium(II)
David J. Bray, Jack K. Clegg, Li-Ling Liao, Leonard F. Lindoy, John C. McMurtrie, David Schilter,
Gang Wei and Tae-Jin Won
S1. Comment
Our group has long been interested in the use of metal complexes as components for the construction of large
supramolecular architectures (Lindoy & Atkinson, 2000). In particular, we are interested in the construction and
chemistry of metallocyclic systems (Clegg et al., 2004, 2005). The title compound, (I), has found extensive use as a
precursor in the preparation of cyclic metallo-supramolecular structures (Fujita et al., 2005). Crystals suitable for this
study were obtained in the course of our investigation into the interactions of N-donor ligand systems (Bray et al., 2005)
with (I).
An ORTEP (Farrugia, 1997) representation of (I) is given in Fig. 1. As expected, each PdII ion has a geometry close to
an ideal square-plane (Table 1). The N donor atoms of the bidentate ethane-1,2-diamine ligand (en) occupy two
coordination sites in a typical five-membered chelate arrangment. The remaining coordination sites are occupied by
nitrate O atoms of two nitrate ligands.
The asymmetric unit contains two of these complexes, which pack via intermolecular hydrogen bonds between the NH2
groups of the en ligands and the O atoms of the nitrate ligands. Hydrogen-bond details are provided in Table 2.
The intermolecular hydrogen bonds propagate in two dimensions, forming an infinite sheet-like motif that lies parallel
to the bc plane (Fig. 2). Each of the N donor atoms forms hydrogen bonds to (at least) two O acceptor atoms, with only
atoms O5 and O8 not involved in close interactions. The sheets stack along the a axis, as shown in the crystal packing
diagram (Fig. 3).
S2. Experimental
The title compound was prepared from cis-[Pd(en)Cl2] and identified as the desired product by comparison with literature
data (Fujita et al., 1996; Tercero-Moreno et al., 1996). Crystals of (I) suitable for the X-ray diffraction study were
isolated from methanol after several days of slow evaporation. All reagents were purchased from Sigma–Aldrich.
S3. Refinement
C-bound H atoms were included in idealized positions and refined using a riding-model approximation, with methylene C
—H bond lengths fixed at 0.99 Å. N-bound H atoms were located in the difference Fourier map and refined with
supporting information
sup-2
[image:5.610.127.482.70.496.2]Acta Cryst. (2005). E61, m1940–m1942 Figure 1
Figure 2
A view of part of one of the two-dimensional sheets formed by hydrogen bonding. The sheets extend infinitely in the bc
supporting information
sup-4
[image:7.610.157.459.70.519.2]Acta Cryst. (2005). E61, m1940–m1942 Figure 3
A view of (I), along the b axis. Alternate two-dimensional sheets are shown in red and green. There are no
hydrogen-bonding interactions connecting adjacent layers. Blue arrows indicate ?
(Ethane-1,2-diamine)dinitratopalladium(II)
Crystal data
C2H8N4O6Pd Mr = 290.52 Monoclinic, P21/c
Hall symbol: -P2ybc a = 16.8478 (6) Å b = 7.7746 (3) Å c = 13.0702 (5) Å β = 109.816 (1)°
V = 1610.62 (10) Å3 Z = 8
F(000) = 1136 Dx = 2.396 Mg m−3
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 10524 reflections θ = 2.4–28.3°
T = 150 K Needle, colourless
0.51 × 0.30 × 0.16 mm
Data collection
Bruker SMART 1000 CCD area-detector diffractometer
Radiation source: sealed tube Graphite monochromator ω scans
Absorption correction: multi-scan (SADABS; Sheldrick, 1999) Tmin = 0.514, Tmax = 0.690
15349 measured reflections 3881 independent reflections 3554 reflections with I > 2σ(I) Rint = 0.019
θmax = 28.3°, θmin = 3.1° h = −22→22
k = −10→10 l = −17→16
Refinement
Refinement on F2
Least-squares matrix: full R[F2 > 2σ(F2)] = 0.016 wR(F2) = 0.043 S = 1.03 3881 reflections 259 parameters 8 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H atoms treated by a mixture of independent and constrained refinement
w = 1/[σ2(F
o2) + (0.0236P)2 + 0.7251P]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max = 0.002
Δρmax = 1.08 e Å−3
Δρmin = −0.40 e Å−3
Special details
Experimental. The crystal was coated in Exxon Paratone N hydrocarbon oil and mounted on a thin mohair fibre attached
to a copper pin. Upon mounting on the diffractometer, the crystal was quenched to 150(K) under a cold nitrogen gas stream supplied by an Oxford Cryosystems Crystream and data were collected at this temperature. 234 standard
reflections were obtained by recollecting the first 50 CCD frames at the end of data collection. They were then used for a decay correction, giving an overall decay of 0.33%.
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full
covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2,
conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used
only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2
are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
supporting information
sup-6
Acta Cryst. (2005). E61, m1940–m1942
C4 0.42840 (10) 0.8026 (2) 0.23646 (14) 0.0201 (3) H7A 0.4702 0.8251 0.2000 0.024* H7B 0.4325 0.6800 0.2582 0.024* N1 0.16543 (9) 0.38548 (19) 0.39314 (12) 0.0183 (3) H1 0.1764 (13) 0.454 (3) 0.4501 (15) 0.027* H2 0.1697 (13) 0.280 (2) 0.4141 (17) 0.027* N2 0.13345 (9) 0.35657 (19) 0.17776 (12) 0.0196 (3) H3 0.1183 (14) 0.447 (2) 0.1329 (17) 0.029* H4 0.1420 (13) 0.265 (2) 0.1422 (17) 0.029* N3 0.31257 (9) 0.38063 (18) 0.13002 (12) 0.0193 (3) N4 0.40206 (9) 0.39355 (17) 0.47206 (12) 0.0189 (3) N5 0.37339 (10) 0.89330 (19) 0.37507 (12) 0.0194 (3) H5 0.3772 (13) 0.969 (3) 0.4257 (16) 0.029* H6 0.3761 (13) 0.791 (2) 0.4044 (17) 0.029* N6 0.34149 (9) 0.84238 (18) 0.16138 (11) 0.0180 (3) H7 0.3219 (12) 0.749 (2) 0.1230 (16) 0.027* H8 0.3493 (14) 0.919 (2) 0.1163 (17) 0.027* N7 0.10544 (9) 0.83412 (18) 0.08766 (11) 0.0199 (3) N8 0.18726 (9) 0.92392 (16) 0.41271 (12) 0.0186 (3) O1 0.28288 (8) 0.23489 (15) 0.12090 (11) 0.0277 (3) O2 0.34835 (9) 0.44032 (19) 0.06988 (11) 0.0310 (3) O3 0.30658 (8) 0.48244 (15) 0.20513 (10) 0.0236 (3) O4 0.33690 (7) 0.49827 (14) 0.43624 (9) 0.0197 (2) O5 0.40964 (8) 0.27554 (15) 0.41420 (11) 0.0260 (3) O6 0.45183 (8) 0.42377 (15) 0.56363 (11) 0.0270 (3) O7 0.10879 (8) 0.72811 (16) 0.15929 (11) 0.0274 (3) O8 0.04821 (8) 0.84143 (17) 0.00052 (11) 0.0300 (3) O9 0.16642 (7) 0.94747 (15) 0.10590 (10) 0.0196 (2) O10 0.21827 (8) 0.78038 (15) 0.43498 (11) 0.0268 (3) O11 0.14757 (8) 0.99788 (17) 0.46451 (10) 0.0286 (3) O12 0.19575 (7) 1.00942 (15) 0.33215 (9) 0.0209 (2) Pd1 0.239096 (8) 0.419159 (14) 0.302404 (10) 0.01465 (4) Pd2 0.266618 (7) 0.911995 (14) 0.246167 (10) 0.01437 (4)
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
N8 0.0196 (7) 0.0197 (7) 0.0163 (7) −0.0032 (5) 0.0059 (6) −0.0010 (5) O1 0.0347 (7) 0.0199 (6) 0.0305 (7) −0.0016 (5) 0.0137 (6) −0.0052 (5) O2 0.0287 (7) 0.0449 (8) 0.0258 (7) 0.0027 (6) 0.0174 (6) 0.0089 (6) O3 0.0315 (7) 0.0203 (6) 0.0252 (6) −0.0074 (5) 0.0177 (5) −0.0049 (5) O4 0.0174 (5) 0.0187 (5) 0.0206 (6) 0.0016 (4) 0.0033 (5) −0.0043 (4) O5 0.0310 (7) 0.0203 (6) 0.0286 (7) 0.0035 (5) 0.0125 (6) −0.0060 (5) O6 0.0240 (7) 0.0294 (7) 0.0223 (6) 0.0010 (5) 0.0008 (5) −0.0024 (5) O7 0.0306 (7) 0.0239 (6) 0.0277 (7) −0.0027 (5) 0.0097 (6) 0.0063 (5) O8 0.0239 (7) 0.0352 (7) 0.0239 (6) 0.0011 (5) −0.0011 (5) −0.0016 (6) O9 0.0180 (6) 0.0219 (5) 0.0183 (6) −0.0011 (5) 0.0055 (5) 0.0039 (5) O10 0.0335 (7) 0.0197 (6) 0.0281 (6) 0.0026 (5) 0.0116 (6) 0.0069 (5) O11 0.0320 (7) 0.0343 (7) 0.0263 (6) −0.0002 (6) 0.0186 (6) −0.0047 (6) O12 0.0272 (6) 0.0196 (6) 0.0196 (6) 0.0043 (5) 0.0129 (5) 0.0036 (5) Pd1 0.01615 (7) 0.01443 (7) 0.01398 (7) −0.00212 (4) 0.00591 (5) −0.00176 (4) Pd2 0.01580 (7) 0.01446 (7) 0.01336 (7) 0.00110 (4) 0.00559 (5) 0.00127 (4)
Geometric parameters (Å, º)
C1—N1 1.482 (2) N3—O2 1.2319 (19) C1—C2 1.505 (2) N3—O3 1.2914 (19) C1—H2A 0.9900 N4—O5 1.2230 (18) C1—H2B 0.9900 N4—O6 1.2289 (19) C2—N2 1.486 (2) N4—O4 1.3185 (17) C2—H3A 0.9900 N5—Pd2 2.0102 (15) C2—H3B 0.9900 N5—H5 0.873 (15) C3—N5 1.489 (2) N5—H6 0.875 (15) C3—C4 1.504 (2) N6—Pd2 2.0150 (14) C3—H6A 0.9900 N6—H7 0.880 (15) C3—H6B 0.9900 N6—H8 0.881 (15) C4—N6 1.493 (2) N7—O8 1.2189 (19) C4—H7A 0.9900 N7—O7 1.2341 (19) C4—H7B 0.9900 N7—O9 1.3126 (18) N1—Pd1 2.0032 (14) N8—O10 1.2248 (18) N1—H1 0.882 (15) N8—O11 1.2422 (19) N1—H2 0.863 (15) N8—O12 1.2935 (18) N2—Pd1 2.0231 (14) O3—Pd1 2.0326 (12) N2—H3 0.897 (15) O4—Pd1 2.0492 (11) N2—H4 0.886 (15) O9—Pd2 2.0465 (12) N3—O1 1.2279 (19) O12—Pd2 2.0426 (11)
supporting information
sup-8
Acta Cryst. (2005). E61, m1940–m1942
C1—C2—H3A 110.1 Pd2—N5—H6 109.3 (14) N2—C2—H3B 110.1 H5—N5—H6 108 (2) C1—C2—H3B 110.1 C4—N6—Pd2 110.58 (10) H3A—C2—H3B 108.4 C4—N6—H7 107.4 (14) N5—C3—C4 106.87 (13) Pd2—N6—H7 110.6 (14) N5—C3—H6A 110.3 C4—N6—H8 104.2 (14) C4—C3—H6A 110.3 Pd2—N6—H8 116.6 (14) N5—C3—H6B 110.3 H7—N6—H8 106.9 (19) C4—C3—H6B 110.3 O8—N7—O7 123.94 (15) H6A—C3—H6B 108.6 O8—N7—O9 117.45 (14) N6—C4—C3 107.43 (13) O7—N7—O9 118.61 (14) N6—C4—H7A 110.2 O10—N8—O11 123.87 (15) C3—C4—H7A 110.2 O10—N8—O12 120.38 (14) N6—C4—H7B 110.2 O11—N8—O12 115.74 (13) C3—C4—H7B 110.2 N3—O3—Pd1 122.12 (10) H7A—C4—H7B 108.5 N4—O4—Pd1 116.72 (9) C1—N1—Pd1 107.22 (11) N7—O9—Pd2 115.43 (9) C1—N1—H1 108.2 (14) N8—O12—Pd2 120.26 (10) Pd1—N1—H1 115.3 (14) N1—Pd1—N2 83.84 (6) C1—N1—H2 108.2 (14) N1—Pd1—O3 172.80 (6) Pd1—N1—H2 108.1 (14) N2—Pd1—O3 94.62 (5) H1—N1—H2 110 (2) N1—Pd1—O4 90.81 (5) C2—N2—Pd1 110.66 (10) N2—Pd1—O4 173.19 (5) C2—N2—H3 108.3 (14) O3—Pd1—O4 90.16 (5) Pd1—N2—H3 108.4 (14) N5—Pd2—N6 83.82 (6) C2—N2—H4 108.0 (14) N5—Pd2—O12 95.14 (5) Pd1—N2—H4 111.1 (14) N6—Pd2—O12 173.68 (5) H3—N2—H4 110 (2) N5—Pd2—O9 173.01 (5) O1—N3—O2 123.85 (15) N6—Pd2—O9 91.35 (5) O1—N3—O3 120.34 (14) O12—Pd2—O9 89.11 (5)
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
N2—H3···O7 0.90 (2) 2.23 (2) 2.9167 (19) 134 (2) N2—H3···O11i 0.90 (2) 2.45 (2) 3.0911 (19) 129 (2)
N2—H4···O1 0.89 (2) 2.49 (2) 3.009 (2) 118 (2) N2—H4···O9ii 0.89 (2) 2.58 (2) 3.415 (2) 158 (2)
N5—H6···O4 0.88 (2) 2.45 (2) 3.2837 (19) 160 (2) N5—H6···O4 0.88 (2) 2.45 (2) 3.2837 (19) 160 (2) N1—H2···O11ii 0.86 (2) 2.35 (2) 3.199 (2) 167 (2)
N1—H2···O12ii 0.86 (2) 2.46 (2) 3.1177 (19) 133 (2)
N1—H1···O9iii 0.88 (2) 2.24 (2) 3.0642 (18) 157 (2)
N6—H7···O3 0.88 (2) 2.39 (2) 2.9550 (18) 122 (2) N6—H7···O10i 0.88 (2) 2.50 (2) 3.1324 (19) 130 (2)
N6—H7···O2 0.88 (2) 2.58 (2) 3.363 (2) 149 (2) N6—H8···O4i 0.88 (2) 2.38 (2) 3.1695 (18) 150 (2)
N6—H8···O6i 0.88 (2) 2.40 (2) 3.1628 (19) 145 (2)