organic papers
Acta Cryst.(2005). E61, o2531–o2533 doi:10.1107/S1600536805021951 Perpe´tuo and Janczak C
4H11N2+NO3
o2531
Acta Crystallographica Section EStructure Reports Online
ISSN 1600-5368
Piperazinium nitrate
Genivaldo Julio Perpe´tuoaand
Jan Janczakb*
a
Departamento de Fisica, Instituto de Cieˆncias Exatas e Biolo´gicas, Universidade Federal de Ouro Preto, CEP 35.400-000 – Ouro Preto – MG, Brazil, andbInstitute of Low Temperature
and Structure Research, Polish Academy of Sciences, PO Box 1410, 50-950 Wrocław, Poland
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study
T= 295 K
Mean(C–C) = 0.002 A˚
Rfactor = 0.043
wRfactor = 0.108
Data-to-parameter ratio = 19.7
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2005 International Union of Crystallography Printed in Great Britain – all rights reserved
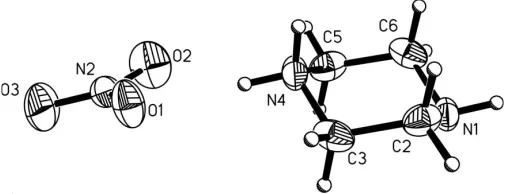
Crystals of the title compound, C4H11N2 +
NO3
, are built up from singly protonated piperazinium residues and nitrate anions. The components are linked by hydrogen bonds into a three-dimensional framework. The piperazinium residues are linked togetherviaN—H N hydrogen bonds into chains in the form of stacks along the [100] direction.
Comment
The present study is a continuation of the work on compounds that form non-covalent supramolecular framework structures in the solid-state viamultiple hydrogen bonds (Perpe´tuo & Janczak, 2004), in order to expand our understanding of the physical–organic chemistry of systems containing multiple N—H N and N—H O hydrogen bonds in the solid state. We present here the solid-state structure of piperazinium nitrate, (I). Selected geometric parameters are given in Table 1.
The singly protonated piperazinium ring in the crystal structure of (I) adopts the chair conformation (Fig. 1) predicted by molecular orbital calculations for the isolated ion (Frisch et al., 1998). As a result of protonation, the C—N bonds involving the protonated N atom are slightly longer than the C—N bonds containing the non-protonated N atom (Table 1). The correlation between the C—N bonds is similar to that found in the gas-phase structure obtained by DFT using the B3LYP method; the optimized C—N bond lengths involving the protonated N atom are 1.510 A˚ , while the C—N
[image:1.610.228.438.371.464.2] [image:1.610.206.460.626.723.2]Received 16 June 2005 Accepted 8 July 2005 Online 16 July 2005
Figure 1
bond lengths involving the non-protonated N atom are 1.445 A˚ .
The geometry of the nitrate anion in (I) shows a slight distortion fromD3hsymmetry obtained by molecular orbital
calculations [the three N—O bonds in the isolated NO
3 ion
[image:2.610.47.294.71.233.2]are equivalent, with a length of 1.226 A˚ (Frisch et al., 1998)] due to the interaction with the piperazinium cation and the formation of hydrogen bonds. The N—O bond lengths in the nitrate anion are indicative of a bond order between 1 and 2, reflecting delocalization of two bonds over three N. . .O bonds. The O atom with the shortest N—O bond length (O3) does not form a hydrogen bond and the other two O atoms are involved in hydrogen bonds as acceptors, one in two hydrogen bonds (O1) and the other in one hydrogen bond (O2) (Table 2).
In the crystal structure, the piperazinium C4H11N
þ
2 cations
are linkedviaN—H N hydrogen bonds into chains, in the form of stacks parallel to the [100] direction (Fig. 2). These chains are interconnectedviaN—H O hydrogen bonds with the nitrate anions into a three-dimensional framework superstructure.
A survey of the Cambridge Structural Database (CSD; Version 5.26; Allen, 2002) for systems containing piperazinium cations yields over 300 structures with the doubly protonated piperazinium(2+) cation and a few structures with a singly protonated piperazinium(+) cation. The singly protonated piperazinium(+) cation mainly forms complexes with metals in which the non-protonated N atom of the piperazinium ring coordinates to the metal, as found in several open-framework structures (Neerajet al., 2001, 2002; Francis & Jacobson, 2001), or forms salts with large organic acids, as found in the struc-ture of piperazinium 5,7-dihydroxy-3-(4-hydroxyphenyl)-4H -1-benzopyran-4-one hydrate (Kozerski et al., 2003). In all structures containing the singly protonated piperazinium(+) cation, the ring, as observed for (I), adopts a chair confor-mation. The structures of piperazinium salts of basic organic
or inorganic acids contain doubly protonated
piper-azinium(2+) cations. Thus, (I) represents the first structurally characterized simple inorganic salt containing a singly protonated piperazinium cation.
Experimental
Piperazine (99%) purchased from Aldrich was dissolved in 10% nitric acid. After several days, colourless single crystals appeared.
Crystal data
C4H11N þ 2NO
3
Mr= 149.16
Monoclinic,P21=c
a= 4.4420 (9) A˚ b= 12.953 (3) A˚ c= 12.677 (3) A˚
= 95.62 (3)
V= 725.9 (3) A˚3
Z= 4
Dx= 1.365 Mg m 3
MoKradiation Cell parameters from 937
reflections
= 3.2–29.5
= 0.12 mm1
T= 295 (2) K
Parallelepiped, colourless 0.420.350.22 mm
Data collection
Kuma KM-4 CCD area-detector diffractometer
!scans
Absorption correction: analytical face-indexed (SHELXTL; Sheldrick, 1990) Tmin= 0.948,Tmax= 0.970
8599 measured reflections
1874 independent reflections 939 reflections withI> 2(I) Rint= 0.018
max= 29.5
h=5!5 k=17!16 l=16!17
Refinement
Refinement onF2
R[F2> 2(F2)] = 0.043 wR(F2) = 0.108
S= 1.05 1874 reflections 95 parameters
H atoms treated by a mixture of independent and constrained refinement
w= 1/[2(F
o2) + (0.0421P)2]
whereP= (Fo2+ 2Fc2)/3
(/)max= 0.003 max= 0.14 e A˚
3
min=0.14 e A˚ 3
[image:2.610.314.566.478.537.2]Extinction correction:SHELXL97 Extinction coefficient: 0.012 (3)
Table 1
Selected geometric parameters (A˚ ,).
N2—O3 1.2114 (15) N2—O2 1.2340 (14) N2—O1 1.2388 (15) N1—C2 1.4445 (18)
N1—C6 1.4592 (19) C3—N4 1.4841 (18) N4—C5 1.4669 (18)
O3—N2—O2 124.63 (14) O3—N2—O1 119.14 (14)
[image:2.610.314.565.598.654.2]O2—N2—O1 116.22 (12)
Table 2
Hydrogen-bond geometry (A˚ ,).
D—H A D—H H A D A D—H A
N4—H41 O1 0.90 2.07 2.9222 (14) 158 N4—H41 O2 0.90 2.32 3.0849 (15) 142 N4—H42 N1i
0.90 1.92 2.8219 (16) 178 N1—H1 O1ii
0.85 (2) 2.27 (2) 3.0486 (15) 153 (1)
Symmetry codes: (i)xþ1;y;z; (ii)x1;yþ1 2;zþ
1 2.
The H atoms bonded to C atoms and N4 were placed in idealized positions and refined as riding atoms, with C—H and N—H distances constrained to 0.97 and 0.90 A˚ , respectively, and Uiso(H) =
1.5Ueq(parent atom). The H atom bonded to N1 was located in a
difference Fourier map and restrained with N—H = 0.85 (1) A˚ and
Uiso(H) = 1.5Ueq(N1).
organic papers
o2532
Perpe´tuo and Janczak C4H11N+2NO3 Acta Cryst.(2005). E61, o2531–o2533
Figure 2
Data collection:KM-4 CCD Software (Kuma, 2001); cell refine-ment: KM-4 CCD Software; data reduction: KM-4 CCD Software; program(s) used to solve structure: SHELXS97 (Sheldrick, 1997); program(s) used to refine structure:SHELXL97(Sheldrick, 1997); molecular graphics:SHELXTL (Sheldrick, 1990); software used to prepare material for publication:SHELXL97.
GJP thanks the CNPq foundation (Brazil) for financial support.
References
Allen, F. H. (2002).Acta Cryst.B58, 380–388.
Francis, R. J. & Jacobson, A. J. (2001).Angew. Chem. Int. Ed.40, 2879–2881.
Frisch, J. M., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., Zakrzewski, V. G., Montgomery Jr, J. A., Stratmann, R. E., Burant, J. C.et al.(1998).GAUSSIAN98. Revision A3. Gaussian Inc., Pittsburgh, PA, USA.
Kozerski, L., Kamien´ski, B., Kawe´cki, R., Urbanczyk-Lipkowska, Z., Bocian, W., Bednarek, E., Sitkowski, J., Zakrzewska, K., Nielsen, K. T. & Hansen, P. E. (2003).Org. Biomol. Chem.1, 3578–3585.
Kuma (2001). KM-4 CCD Software. Version 171.1. Kuma Diffraction, Wrocław, Poland.
Neeraj, S., Forster, P. M., Rao, C. N. R. & Cheetham, A. K. (2001).Chem. Commun.pp. 2716–2717.
Neeraj, S., Noy, M. L., Rao, C. N. R. & Cheetham, A. N. (2002).J. Solid State Chem.167, 344–353.
Perpe´tuo, G. J. & Janczak, J. (2004).Acta Cryst.C60, o768–o770.
Sheldrick, G. M. (1990).SHELXTL.Siemens Analytical X-ray Instruments Inc., Madison, Wisconsin, USA.
Sheldrick, G. M. (1997).SHELXS97andSHELXL97. University of Go¨tingen, Germany.
organic papers
Acta Cryst.(2005). E61, o2531–o2533 Perpe´tuo and Janczak C
supporting information
sup-1 Acta Cryst. (2005). E61, o2531–o2533
supporting information
Acta Cryst. (2005). E61, o2531–o2533 [https://doi.org/10.1107/S1600536805021951]
Piperazinium nitrate
Genivaldo Julio Perp
é
tuo and Jan Janczak
(I)
Crystal data
C4H11N2+·NO3− Mr = 149.16
Monoclinic, P21/c Hall symbol: -P 2ybc a = 4.4420 (9) Å b = 12.953 (3) Å c = 12.677 (3) Å β = 95.62 (3)° V = 725.9 (3) Å3 Z = 4
F(000) = 320
Dx = 1.365 Mg m−3 Dm = 1.36 Mg m−3 Dm measured by flotation Mo Kα radiation, λ = 0.71073 Å Cell parameters from 937 reflections θ = 3.2–29.5°
µ = 0.12 mm−1 T = 295 K
Parallelepiped, colourless 0.42 × 0.35 × 0.22 mm
Data collection
Kuma KM-4 with CCD area-detector diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
Detector resolution: 1024x1024 with blocks 2x2 pixels mm-1
ω scans
Absorption correction: analytical
face-indexed (SHELXTL; Sheldrick, 1990)
Tmin = 0.948, Tmax = 0.970 8599 measured reflections 1874 independent reflections 939 reflections with I > 2σ(I) Rint = 0.018
θmax = 29.5°, θmin = 3.2° h = −5→5
k = −17→16 l = −16→17
Refinement
Refinement on F2 Least-squares matrix: full R[F2 > 2σ(F2)] = 0.043 wR(F2) = 0.108 S = 1.05 1874 reflections 95 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H atoms treated by a mixture of independent and constrained refinement
w = 1/[σ2(F
o2) + (0.0421P)2] where P = (Fo2 + 2Fc2)/3 (Δ/σ)max = 0.003
Δρmax = 0.14 e Å−3 Δρmin = −0.14 e Å−3
supporting information
sup-2 Acta Cryst. (2005). E61, o2531–o2533
Special details
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
N2 0.9850 (2) 0.04477 (10) 0.26449 (9) 0.0658 (4) O1 0.9832 (2) 0.14018 (8) 0.25769 (7) 0.0678 (4) O2 0.9157 (3) 0.00830 (8) 0.34873 (10) 0.0794 (5) O3 1.0575 (3) −0.00589 (10) 0.19074 (12) 0.0913 (5) N1 0.2485 (2) 0.29227 (10) 0.55572 (9) 0.0551 (4) H1 0.201 (3) 0.3301 (11) 0.6059 (12) 0.083* C2 0.4173 (3) 0.35955 (10) 0.49228 (12) 0.0605 (4) H21 0.5880 0.3887 0.5358 0.091* H22 0.2891 0.4159 0.4645 0.091* C3 0.5269 (3) 0.30026 (12) 0.40327 (10) 0.0612 (4) H31 0.3558 0.2746 0.3573 0.092* H32 0.6452 0.3449 0.3618 0.092* N4 0.7157 (2) 0.21240 (9) 0.44595 (9) 0.0595 (4) H41 0.7705 0.1741 0.3917 0.089* H42 0.8851 0.2369 0.4821 0.089* C5 0.5545 (3) 0.14682 (10) 0.51619 (12) 0.0634 (4) H51 0.6891 0.0934 0.5468 0.095* H52 0.3847 0.1136 0.4758 0.095* C6 0.4442 (3) 0.21019 (12) 0.60193 (10) 0.0611 (4) H61 0.3326 0.1670 0.6470 0.092* H62 0.6147 0.2400 0.6451 0.092*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
supporting information
sup-3 Acta Cryst. (2005). E61, o2531–o2533
Geometric parameters (Å, º)
N2—O3 1.2114 (15) C3—H31 0.9700 N2—O2 1.2340 (14) C3—H32 0.9700 N2—O1 1.2388 (15) N4—C5 1.4669 (18) N1—C2 1.4445 (18) N4—H41 0.9000 N1—C6 1.4592 (19) N4—H42 0.9000 N1—H1 0.846 (15) C5—C6 1.483 (2) C2—C3 1.486 (2) C5—H51 0.9700 C2—H21 0.9700 C5—H52 0.9700 C2—H22 0.9700 C6—H61 0.9700 C3—N4 1.4841 (18) C6—H62 0.9700
O3—N2—O2 124.63 (14) C5—N4—C3 111.92 (10) O3—N2—O1 119.14 (14) C5—N4—H41 109.2 O2—N2—O1 116.22 (12) C3—N4—H41 109.2 C2—N1—C6 109.99 (10) C5—N4—H42 109.2 C2—N1—H1 104.5 (10) C3—N4—H42 109.2 C6—N1—H1 107.6 (10) H41—N4—H42 107.9 N1—C2—C3 109.72 (12) N4—C5—C6 109.90 (11) N1—C2—H21 109.7 N4—C5—H51 109.7 C3—C2—H21 109.7 C6—C5—H51 109.7 N1—C2—H22 109.7 N4—C5—H52 109.7 C3—C2—H22 109.7 C6—C5—H52 109.7 H21—C2—H22 108.2 H51—C5—H52 108.2 N4—C3—C2 109.57 (11) N1—C6—C5 109.61 (11) N4—C3—H31 109.8 N1—C6—H61 109.7 C2—C3—H31 109.8 C5—C6—H61 109.7 N4—C3—H32 109.8 N1—C6—H62 109.7 C2—C3—H32 109.8 C5—C6—H62 109.7 H31—C3—H32 108.2 H61—C6—H62 108.2
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
N4—H41···O1 0.90 2.07 2.9222 (14) 158 N4—H41···O2 0.90 2.32 3.0849 (15) 142 N4—H42···N1i 0.90 1.92 2.8219 (16) 178 N1—H1···O1ii 0.846 (15) 2.270 (15) 3.0486 (15) 153.2 (12)
![Figure 2A view of the crystal packing, showing the hydrogen-bonded N—H� � �Nchains (dashed lines) along the [100] direction.](https://thumb-us.123doks.com/thumbv2/123dok_us/706275.574182/2.610.47.294.71.233/figure-crystal-packing-showing-hydrogen-bonded-nchains-direction.webp)
![catena Poly[[(diaquazinc) μ 3 carboxypyrazine 2 carboxylato κ4N1,O2;N4,O3] nitrate]](data:image/gif;base64,R0lGODlhAQABAIAAAP///wAAACH5BAEAAAAALAAAAAABAAEAAAICRAEAOw==)