Mimics of pincer ligands: an accessible phosphine-free
N-(pyrimidin-2-yl)-1,2-azole-3-carboxamide framework for binuclear Pd (II) complexes and
high-turnover catalysis in water
Alexey V. Kletskov,*a,b,c Nikolay A. Bumagin,*a Sergey K. Petkevich,b Evgenij A. Dikusar,b Alexander S. Lyakhov, d Ludmila S. Ivashkevich,d Iryna A. Kolesnik,b Vladimir I. Potkin*b
a M.V. Lomonosov Moscow State University, Chemical Department, Leninskie Gory, 1/3, Moscow
119991, Russian Federation. E-mail: [email protected],
b Institute of Physical Organic Chemistry, National Academy of Sciences of Belarus, Surganova Str.,
13, Minsk 220072, Belarus.E-mail: [email protected]
cPeoples’ Friendship University of Russia (RUDN University), Moscow, Miklukho-Maklaya Str., 6,
117198, Russian Federation. E-mail: [email protected]
d Research Institute for Physical Chemical Problems of Belarusian State University, Leningradskaya
Str., 14, Minsk 220006, Belarus.
SUPPORTING INFORMATION
Experimental part...1
General Procedure for the Synthesis of N-(pyrimidin-2-yl)isoxazole(isothiazole)-3-carboxamides 3–6 ...2
Procedure for the Synthesis of Pd complexes Pd2Cl2(L-H)2 7-10...9
X-Ray data for complexes 8 and 10 ...12
General procedure for the Suzuki-Miyaura reaction...14
General procedure for the Suzuki-Miyaura reaction of aryl bromides with arylboronic acids bearing electron-withdrawing groups...19
General procedure for the Mizoroki–Heck reaction...33
General procedure for the Sonogashira reaction...36
Experimental part
The IR spectra were recorded on a FTIR spectrometer Protege-460 (Nicolet) in KBr pellets.1H, and 13C NMR spectra were recorded on a Bruker Avance-500 or Bruker Avance-II NMR 400 spectrometers operating at 500, 400 and 125, 100 MHz for 1H and 13C, respectively. Chemical shifts are given on the δ scale (ppm) and were determined relative to the residual solvent signal (δН 7.25 ppm, δС 77.0 ppm for CDCl3; δН 2.5 ppm, δС 40.1 ppm for DMSO-d6, δН 8.0 ppm, δС 162.5 ppm for DMF-d7). Coupling constants (J) are given in Hertz. Mass spectra were obtained on an instrument Agilent 5975 inert MSD/6890N Network GC System (EI ionisation at 70 eV), HP-5MS capillary column 30 m × 0.25 mm, 5% PhMe Silicon stationary phase (0.25 μm), evaporator temperature 250
°С. Data are reported as m/z (relative intensity in %). HPLC-MS studies were performed using Agilent 6470 and Agilent 1200 liquid chromatographs with mass-selective detector Agilent 6410 Triple Quad in Positive ESI mode. Elemental analysis was performed on a Vario Micro cube CHNS-analyser. The chlorine content was determined by classical microanalysis according to the modified Pregl method. The palladium content of complexes and residual palladium contamination in target products was determined by atomic absorption spectroscopy on a MGA-915 spectrometer. Microscope Levenhuk Rainbow D50L PLUS (x64–1280) was used for monitoring presence of Pd-black. TLC was performed on Merck Silica gel 60 F254 plates; the eluent was hexane–Et2O, 2:1-1:3. Melting points were determined on Boetius apparatus. All purchased chemicals were used as such.
General Procedure for the Synthesis of
N-(pyrimidin-2-yl)isoxazole(isothiazole)-3-carboxamides3–6
To a solution of pyrimidine or 4,6-dimethylpyrimidine (2.4 mmol) in anhydrous pyridine (50 mL), 2.4 mmol 5-phenylisoxazol-3-carbonyl chloride or 4,5-dichloroisothiazol-3-carbonyl chloride was added and the mixture was stirred at room temperature for 3 h. The precipitate was filtered off, washed with 10% aqueous sodium bicarbonate, water (3×25mL) and dried in vacuo over P2O5. The obtained amides don’t need further purification.
5-Phenyl N-(pyrimidin-2-yl)isoxazole-3-carboxamide 3. White solid (88 % yield); m.p.
205–206 C. 1H NMR (500 MHz, DMSO-d6): δ = 7.32 (s, 1Н
isox.), 7.48–7.62 (m, 3Нarom.+
1Нpyrim.), 7.90–8.03 (m, 2Нarom.), 8.72–8.82 (m, 2Hpyrim.), 11.11 (s, 1Н, NH) ppm. 13C NMR (125 MHz, DMSO-d6): δ = 100.76 (CHisox.), 118.64 (1CHpyrim.), 126.40 (2CHarom.), 129.97 (2CHarom.), 131.55 (1CHarom.), 159.22 (2CHpyrim.), 126.82, 157.64, 157.96, 160.22, 171.14 (5Cquat.), 168.47 (2Cquat.) ppm. IR (KBr): ṽmax= 3359, 3148, 3129, 3058; 1708, 1613, 1580, 1570, 1514, 1497, 1454, 1429, 1408, 1323, 1219, 1129, 1007, 948, 895, 844, 822, 794, 764, 688, 673, 654, 637, 629, 612, 517 cm–1. GC-MS (EI) m/z (I, %): 266 (16) [M]+, 238 (29), 189 (33), 161 (16), 122 (100), 105 (68), 79 (100). Anal. Calcd for C14H10N4O2: C 63.15; H 3.79; N 21.04. Found C 63.31; H 3.89; N 20.97.
1H NMR of 3
MS of 3
N-(4,6-dimethylpyrimidin-2-yl)-5-phenylisoxazole-3-carboxamide 4. White solid (87 %
yield); m.p. 156 C. 1H NMR (500 MHz, DMSO-d6): δ= 2.38 (s, 6Н, 2CH
3), 7.05 (s, 1Нpyrim.), 7.48 (s, СНisox.), 7.53–7.59 (m, 3Нarom.), 7.91–7.98 (m, 2Нarom.), 10.83 (s, 1Н, NH) ppm. 13C NMR (125 MHz, DMSO-d6): δ = 23.90 (2CH3), 100.62 (CH3), 117.18 (CHpyrim.), 126.35 (2CHarom.), 129.93 (2CHarom.), 131.47 (1CHarom.), 126.85, 157.07, 157.91, 160.28, 170.98 (5Cquat.), 168.47 (2Cquat.) ppm. IR (KBr): ṽmax= 3146, 3130, 3063, 3038, 2991, 2960, 2921, 2850; 1728, 1605, 1580, 1570, 1524, 1452, 1427, 1386, 1346, 1232, 1193, 1141, 969, 947, 859, 783, 774, 763, 700, 691, 636, 597, 544 cm–1. GC-MS (EI) m/z (I, %): 294 (57) [M]+, 265 (22), 217 (57), 189 (20), 150 (100), 105 (91), 77 (65). Anal. Calcd for C16H14N4O2: C 65.30; H 4.79; N 19.04. Found C 65.42; H 4.87; N 18.98.
1H NMR of 4
MS of 4
4,5-Dichloro-N-(pyrimidin-2-yl)isothiazole-3-carboxamide 5. White solid (83 % yield);
m.p. 146–148 C. 1H NMR (500 MHz, DMSO-d6): δ = 7.25 (t, 1Н
pyrim., J = 4.8), 8.66 (d,
2Нpyrim., J = 4.9), 11.31 (s, 1Н, NH) ppm. 13C NMR (125 MHz, DMSO-d6): δ = 118.23 (CHpyrim.), 159.84 (2CHpyrim.), 122.62, 149.33, 157.49, 159.06, 159.51 (5Cquat.) ppm. IR (KBr):
ṽmax= 3282, 3110, 3061; 1722; 1585, 1569, 1508, 1475, 1434, 1414, 1347, 1327, 1194, 1164, 1086, 1073, 976, 893, 858, 831, 817, 793, 752, 634, 609, 505 cm–1. HPLC-MS, m/z (I, %): 274.9 [M + H ]+, calcd 274.96, (100). Anal. Calcd for C
8H4Cl2N4OS: C 34.93; H 1.47; Cl 25.77; N 20.37; S 11.65. Found C 34.81; H 1.68; Cl 25.61; N 20.43; S 11.59.
1H NMR of 5
MS of 5
4,5-Dichloro-N-(4,6-dimethylpyrimidin-2-yl)isothiazole-3-carboxamide 6. White solid (84
% yield); m.p. 176–178 C. 1H NMR (500 MHz, DMSO-d6): δ = 2.25 (s, 6Н, 2CH
3), 6.96 (s,
1Нpyrim.), 11.25 (s, 1Н, NH) ppm. 13C NMR (125 MHz, DMSO-d6): δ = 23.82 (2CH3), 116.57 (CHpyrim.), 122.33, 148.46, 160.80, 161.13, 168.27 (5Cquat.), 156.96 (2Cquat.) ppm. IR (KBr):
ṽmax= 3328, 2986, 2924, 2851, 1720, 1603, 1530, 1479, 1439, 1384, 1347, 1225, 1183, 1176, 1099, 1037, 874, 859, 786, 764, 690, 646, 607, 510 cm–1. HPLC-MS, m/z (I, %): 303.0 [M + H
]+, calcd 302.99, (100). Anal. Calcd for C
10H8Cl2N4OS: C 39.62; H 2.66; Cl 23.39; N 18.48; S 10.58. Found C 39.49; H 2.89; Cl 23.22; N 18.39; S 10.48.
13C NMR of 6
pressure to 1/3 of the initial volume and then the precipitated product was filtered and treated in similar manner.
Complex Pd2Cl2(L1-H)2 7. Light brown solid (97 % yield).IR (KBr): ṽmax= 3236, 3193,
3138, 3083, 3039, 1663, 1605, 1579, 1557, 1500, 1478, 1464, 1412, 1351, 1265, 1235, 1121, 1014, 954, 950, 850, 814, 772, 764, 695, 643, 561. Anal. Calcd for C28H18Cl2N8O4Pd2: C 41.30; H 2.23; Cl 8.71; N 13.76; Pd 24.14. Found C 41.49; H 2.48; Cl 8.55; N 13.87; Pd 24.21.
Complex Pd2Cl2(L2-H)2 8. Light brown solid (92 % yield). The complex was either insoluble
in common solvents for NMR or decomposed due to ligand exchange. Herein we give most
intense peaks for 13C NMR of the complex in DMF-d
7. 13C NMR (125 MHz, DMF-d7): δ =
23.25 (CH3), 23.33 (CH3), 25.00 (CH3), 26.53 (CH3), 100.26 (CHisox.), 100.67 (CHisox.), 118.29 (CHpyrim.), 118.88 (CHpyrim.), 126.52, 126.86 (4СНarom.), 129.80, 129.96 (4СНarom.), 131.52, 132.80 (2СНarom.), 125.78, 126.98, 155.35, 155.95, 159.76, 160.48, 162.70, 163.48, 169.46, 170.11, 170.78, 172.61, 172.69, 173.58 (14Сquat.). IR (KBr): ṽmax=: 3263, 3099, 3050, 2923, 2853, 1710, 1619, 1598, 1579, 1549, 1510, 1487, 1446, 1416, 1321, 1271, 1226, 1187, 1114, 1065, 1043, 1023, 1000, 846, 897, 847, 800, 759, 682, 651, 526. HRMS-ESI, m/z: 441.0339 (calcd for [M – 2Cl+2CH3CN]2+ 441.0343), 886.0391(calcd for [M – 2Cl+ CH3CN+CHO2]+ 886.0403). Anal. Calcd for C32H26Cl2N8O4Pd2: C 44.16; H 3.01; Cl 8.15; N 12.87; Pd 24.45. Found C 44.29; H 3.22; Cl 8.24; N 12.93; Pd 24.62. 13C NMR of 8 180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20 Chemical Shift (ppm) 0.01 0.02 0.03 0.04 0.05 0.06 0.07 0.08 0.09 0.10 0.11 0.12 In te ns ity Dimethylformamide-d7 Dimethylformamide-d7 17 2. 81 17 0. 98 17 0. 30 16 3. 68 15 9. 96 15 6. 15 15 5. 55 13 1. 71 13 0. 00 12 6. 72 11 9. 08 10 0. 46 26 .7 3 23.5 3 O N O N N N CH3 CH3 Pd Cl
Complex Pd2Cl2(L3-H)2 9. Red-brown solid (98 % yield). IR (KBr): ṽmax= 3284, 3232, 3190, 3065, 1735, 1663, 1581, 1562, 1503, 1433, 1413, 1360, 1231, 1105, 994, 962, 858, 812, 584. Anal. Calcd for C16H6Cl6N8O2Pd2S2: C 23.10; H 0.73; Cl 25.57; N 13.47; Pd 25.58; S 7.71. Found C 23.36; H 1.12; Cl 25.42; N 13.55; Pd 25.69; S 7.56.
Complex Pd2Cl2(L4-H)2 10. Ruby red crystalls (94 % yield). 1H NMR (500 MHz, CDCl3): δ
= 2.46 (s, 3H, CH3), 2.93 (s, 3H, CH3), 6.67 (s, 1Нpyrim.). 13C NMR (125 MHz, CDCl3): δ = 23.96 (CH3), 25.47 (CH3), 151.33 (CHpyrim.), 117.43, 126.29, 160.63, 162.58, 163.59, 168.67, 171.87 (7Сquat.). IR (KBr): ṽmax= 3218, 3080, 2988, 2893, 2839, 1740, 1651, 1630, 1583, 1529, 1502, 1439, 1381, 1365, 1300, 1265, 1213, 1194, 1113, 1030, 995, 943, 893, 839, 793, 773, 714, 636, 590. HRMS-ESI, m/z: 449.9001 (calcd for [M – 2Cl+2CH3CN]2+ 449.9005), 903.7712 (calcd for [M – 2Cl+ CH3CN+CHO2]+ 903.7726). Anal. Calcd for C20H14Cl6N8O2Pd2S2: C 27.05; H 1.59; Cl 23.95; N 12.62; Pd 23.97; S 7.22. Found C 27.18; H 1.90; Cl 23.81; N 12.71; Pd 23.67; S 7.08. 1H NMR of 8 10 9 8 7 6 5 4 3 2 1 Chemical Shift (ppm) 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1.0 In te ns ity 3.07 3.00 1.00 Chloroform-d 7. 25 6. 67 2. 93 2.46 S N Cl Cl O N Pd Cl N N CH3 CH3
13C NMR of 10 180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20 Chemical Shift (ppm) 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1.0 In te ns ity Chloroform-d Chloroform-d 17 1. 87 16 8. 67 16 3. 59 16 2. 58 16 0. 63 15 1. 33 12 6. 29 117. 43 77 .3 2 77 .0 0 76 .6 8 25 .4 7 23 .9 6 S N Cl Cl O N Pd Cl N N CH3 CH3
X-Ray data for complexes 8 and 10
X-ray diffraction data for complexes 8 and 10 (figure 1) were collected on a SMART Apex II diffractometer using graphite monochromated Mo-Kα radiation (λ = 0.71073 Å). The structures were solved by direct methods (SIR2014)1 and refined on F2 by the full-matrix least squares technique (SHELXL 2014).2 The intensities were corrected for absorption. For both compounds, non-hydrogen atoms were refined anisotropically. All H atoms were placed in calculated positions and refined in a “riding” model, with Uiso(H) = 1.5Ueq(C) for the methyl H atoms, and Uiso(H) = 1.2Ueq(C) for C–H groups. Molecular graphics was performed with the program PLATON.3 Complexes 8 and 10 crystallize in the space groups P and P21/c, respectively. In both compounds there are two palladium and two chlorine atoms, and two ligand molecules in their asymmetric units, all atoms being in general positions (Fig. 1).
Figure 1 Binuclear molecules of complexes Pd2Cl2(L2-H)2 8 (left) and Pd2Cl2(L4-H)2 10 (rigth), with the atom numbering for coordination environment of Pd1 and Pd2 atoms. All hydrogen atoms are omitted for clarity. Colour scheme for atoms: Pd – green, Cl –orange, N – blue, C – black, O – red, S – violet.
Table 1S. Main crystal data and structure refinement details for complexes 8 and 10 Complex 8 Complex 10
Formula C32H26Cl2N8O4Pd2 C20H14Cl6N8O2Pd2S2
Formula weight 870.31 888.01
T (K) 100(2) 100(2)
Crystal system Triclinic Monoclinic
Space group P–1 P21/c Crystal size (mm) 0.410.200.60 0.22 0.20 0.18 a (Å) 10.58710(10) 11.98510(10) b (Å) 10.91880(10) 14.7332(2) c (Å) 15.6583(2) 16.4161(2) () 107.1140(5) 90 () 96.5039(6) 93.6593(4) () 94.7049(6) 90 V (Å3) 1706.06(3) 2892.83(6) Z 2 4 Dc (g cm–3 ) 1.694 2.039 (mm–1) 1.260 1.979 Reflections collected 38180 72424
Independent reflections 9513[Rint = 0.0172] 14080 [R(int) = 0.0222] Restraints 0 0 Parameters 437 365 R1/wR2 [I>2σ(I)] 0.0214/0.0519 0.0178/0.0392 R1/wR2[alldata] 0.0246/0.0537 0.0221/0.0403 Goodness-of-fit 1.058 1.046 CCDC 1976251 1976252
Table 2S. Coordination bond lengths (Ǻ) in complexes 8 and 10 Complex 8 Complex 10 Pd1–N12 (isoxazole/isothiazole) 1.9890(13) 1.9932(8) Pd1–N17 (amide) 2.0331(13) 2.0273(8) Pd1–N29 (pyrimidine) 2.0350(13) 2.0202(8) Pd1–Cl1 2.2844(4) 2.2917(3) Pd2–N22 (isoxazole/isothiazole) 2.0043(13) 1.9863(8) Pd2–N27 (amide) 2.0282(13) 2.0254(8) Pd2–N19 (pyrimidine) 2.0166(13) 2.0199(8) Pd2–Cl2 2.2857(4) 2.2896(3)
General procedure for the Suzuki-Miyaura reaction
A 20 mL Schlenk tube with a magnetic stir bar was charged with 3-bromobenzoic acid 11 (0.5 mmol), 4-methoxyphenylboronic acid 12 (0.6 mmol), K2CO3 (1.25 mmol), 5 ml of H2O and 50 μL of
solution (510-3-510-6 M) of palladium complexes Pd
2Cl2(L-H)2 in MeOH under air atmosphere. The reaction mixture was stirred for the appropriate time and under conditions required (Tables 1). After this time, the mixture was cooled, diluted with 5 mL of H2O, 5 mL of Et2O and acidified by 1 M HCl. The organic phase was separated, and the aqueous layer was extracted with Et2O (2 x 5 mL). The combined organic layers were washed with brine (5 mL), dried over Na2SO4, concentrated in vacuo and the yield was determined by 1H NMR analysis with 1,1,2,2-tetrachloroethane (0.5 mmol) as internal standard (Tables 1, main text) after flush chromatography on silica gel (ether-hexane = 1:3) to remove 4-methoxyphenylboronic acid. The isolated product was obtained by a simple filtration of ether solution through silica gel pad and evaporation of a solvent. At last, 0.111 g of 4-methoxybiphenyl-3-carboxylic acid 13 with a 97 % yield was obtained and high purity (> 98%) was confirmed by elemental analysis.
1H spectrum (400 MHz, DMSO-d
1H spectrum (400 MHz, DMSO-d
6) of reaction mixture for entry 8, Table 1 (main text)
1H spectrum (400 MHz, DMSO-d
1H spectrum (400 MHz, DMSO-d
6) of reaction mixture for entry 11, Table 1 (main text)
1H spectrum (400 MHz, DMSO-d
1H spectrum (400 MHz, DMSO-d
6) of 3-bromobenzoic acid
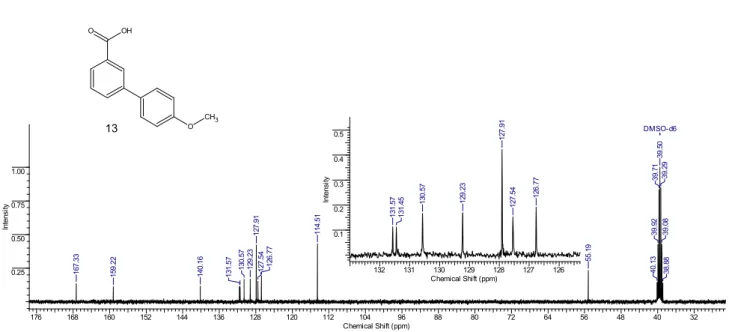
4-Methoxybiphenyl-3-carboxylic acid 13.4 White crystalline powder, m.p. 203-204 °C (m.p. 202–
203 °C4). 1H NMR (400 MHz, DMSO-d 6): δ= 3.79 (s, 3H), 7.03 (d, 2H, J = 8.8), 7.54 (t, 1H, J = 7.8), 7.63 (d, 2H, J = 8.8), 7.82—7.90 (m, 2H), 8.12 (s, 1H). 13C NMR (100 MHz, DMSO-d 6): δ = 55.2, 114.5, 126.8, 127.5, 127.9, 129.2, 130.6, 131.4, 131.6, 140.2, 159.2, 167.3. IR (KBr): 3394, 3081, 2957, 2840, 1678, 1600, 1586, 1423, 1395, 1317, 1289, 1272, 1241, 1197, 1172, 1124, 1062, 1009, 931, 756 cm-1. Calcd for C 14H12O3: C 73.67; H 5.30. Found: C 73.62; H 5.37. 1H NMR of 13 8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 Chemical Shift (ppm) 0 0.05 0.10 0.15 0.20 In te ns ity 3.02 2.01 2.01 1.96 0.99 DMSO-d6 M05 M00 M01 M03 M02 M04 2. 49 2. 49 3. 79 7. 02 7. 04 7. 53 7. 54 7. 61 7. 64 7. 84 7. 84 7. 86 7. 86 7. 88 8. 12 8. 13 O CH3 O OH 13 8.0 7.5 7.0 Chemical Shift (ppm) 0 0.05 0.10 0.15 0.20 In te ns ity 2.01 2.01 1.01 1.96 0.99 M05 M01 M03 M02 M04 8. 13 8. 12 8. 12 7. 88 7. 86 7. 86 7. 85 7.84 7.84 7. 64 7.61 7. 56 7. 54 7. 53 7. 04 7. 02
13C NMR of 13 O OH O CH3 176 168 160 152 144 136 128 120 112 104 96 88 80 72 64 56 48 40 32 Chemical Shift (ppm) 0.25 0.50 0.75 1.00 In te ns ity DMSO-d6 38 .8 8 39 .0 8 39 .2 9 39 .5 0 39 .7 1 39 .9 2 40 .1 3 55 .1 9 11 4. 51 12 6. 77 12 7. 54 12 7. 91 12 9. 23 13 0. 57 13 1. 57 14 0. 16 15 9. 22 16 7. 33 132 131 130 129 128 127 126 Chemical Shift (ppm) 0.1 0.2 0.3 0.4 0.5 In te ns ity 13 1. 57 13 1. 45 130. 57 12 9. 23 12 7. 91 12 7. 54 126. 77 13
General procedure for the Suzuki-Miyaura reaction of aryl bromides with arylboronic acids bearing electron-withdrawing groups
A 50 mL Schlenk tube with a magnetic stir bar was charged with aryl bromides 11a-f (1 mmol), arylboronic acids 12a-g (1.2 mmol), K2CO3 (2.5 mmol), 0.0032 g of Bu4NBr (0.01 mmol, 1 mol%) for water insoluble aryl bromides, 10 ml of H2O and 100 μL of solution (510-4M) of palladium complexes Pd2Cl2(L2-H)2 (0.01 mol% Pd) in MeOH under air atmosphere. The reaction mixture was stirred for the appropriate time and under conditions required (Tables 3S). After this time, the mixture was cooled, diluted with 10 mL of H2O, 10 mL of Et2O and acidified by 1 M HCl (for compounds 13a, 13d-f). The organic phase was separated, and the aqueous layer was extracted with Et2O (2 x 10 mL). The combined organic layers were washed with brine (5 mL), dried over Na2SO4 and concentrated in vacuo to give crude products. The pure products were obtained by a simple filtration of ether-hexane solution through silica gel pad and evaporation of a solvent (Tables 3S).
Table S3. Suzuki-Miyaura reactions of aryl bromides with arylboronic acids bearing electron-withdrawing groups in the presence of complexes Pd2Cl2(L2-H)2a
0.01 mol% "Pd" (1 mol% Bu4NBr ) K2CO3, H2O, 100oC
+
11b 11b 3 c 4-MeOC6H4Br 11c 4-OHCC6H4B(OH)2 11c 10 13cc 97 4 4-HO2CC6H4Br 11d 4-OHCC6H4B(OH)2 11c 5 13dc 93 5 4-HO2CC6H4Br 11d 2-OHCC6H4B(OH)2 11d 8 13dd 95 6 4-HO2CC6H4Br 11d 4-CF3C6H4B(OH)2 11e 5 13de 98 7 c 4-MeOC6H4Br 11c 4-NO2C6H4B(OH)2 11f 10 13cf 97
a Aryl halide (1 mmol), arylboronic acid (1.2 mmol), K
2CO3 (2.5 mmol), 10 mL of H2O, 100 °С.
bIsolated yield.
c In the presence of Bu
4NBr (1 mol%).
4'-Chlorobiphenyl-4-carboxylic acid 13aa.5 Yield 0.22 g (94 %).White crystalline solid, m.p.
292-294 °C (m.p. 293-292-294 °C5). 1H NMR (400 MHz, DMSO-d
6): δ= 7.54 (d, J = 8.4, 2H), 7.75 (d, J = 8.4, 2H), 7.78 (d, J = 8.2, 2H), 8.01 (d, J = 8.2, 2H), 13.02 (br s, 1H). 13C NMR (100 MHz, DMSO-d
6): δ= 126.8, 128.7, 129.0, 130.00, 130.03, 133.2, 137.8, 142.9, 167.1. Calcd for C13H9ClO2: C 67.11; H 3.90, Cl 15.24. Found: C 67.03; H 3.98, Cl 15.20. 1H NMR of 13aa O OH Cl 13 12 11 10 9 8 7 6 5 4 3 2 Chemical Shift (ppm) 4.04 0.97 DMSO-d6 M02 M00 M01 7 .5 2 7 .5 5 7 .7 4 7 .7 9 8 .0 0 8 .0 2 1 3. 0 2 8.5 8.0 7.5 Chemical Shift (ppm) 0 0.25 0.50 0.75 1.00 In te n si ty 4.04 2.00 1.99 M02 M01 M00 8 .0 2 8.0 0 7 .7 9 7 .7 4 7 .5 5 7 .5 2
13C NMR of 13aa Cl O OH 168 160 152 144 136 128 120 112 104 96 88 80 72 64 56 48 40 Chemical Shift (ppm) DMSO-d6 12 6. 78 12 8. 74 12 9. 04 13 0. 00 13 3. 21 13 7. 83 14 2. 87 16 7. 08 134 133 132 131 130 129 128 127 126 Chemical Shift (ppm) 0.1 0.2 0.3 0.4 0.5 In te ns ity 13 3. 21 13 0. 03 13 0. 00 129. 04 12 8. 74 12 6. 78
Methyl 4'-aminobiphenyl-3-carboxylate 13bb. Yield 0.22 g (96 %). Light brownish crystals, m.p.
126-128 °C (decomp.). 1H NMR (400 MHz, DMSO-d
6): δ= 3.86 (s, 3 H) 5.51 (br.s, 2H), 6.69 (d, J = 8.3, 2H), 7.40 (d, J = 8.3, 2H) 7.50 (t, J = 7.7, 1H) 7.79 (m, 2H) 8.07 (s, 1H). 13C NMR (100 MHz, DMSO-d6): δ = 52.1, 114.5, 125.7, 126.3, 126.4, 127.3, 129.2, 130.08, 130.12, 141.1, 148.4, 166.4. Calcd for C14H13NO2: C 73.99; H 5.77, N 6.16. Found: C 73.92; H 5.85, N 6.09.
1H NMR of 13bb N H2 O O CH3 M00 M06 M03 M02 M04 7. 41 7. 39 6. 70 6. 68 3. 86 8.0 7.5 7.0 Chemical Shift (ppm) 0 0.1 0.2 0.3 0.4 0.5 In te ns ity 2.01 1.091.95 2.00 1.00 M06 M03 M05 M02 M04 6. 68 6. 70 7. 39 7. 41 7. 48 7. 50 7. 52 7. 79 8. 07
13C NMR of 13bb N H2 O O CH3 168 160 152 144 136 128 120 112 104 96 88 80 72 64 56 48 40 Chemical Shift (ppm) DMSO-d6 16 6. 38 14 8. 45 14 1. 11 13 0. 08 12 9. 22 12 7. 31 12 5. 71 11 4. 53 52 .1 4 40 .1 2 39 .9 2 39 .7 1 39 .5 0 39 .2 9 39 .0 8 38 .8 8 132 130 128 126 124 Chemical Shift (ppm) 0.25 0.50 0.75 1.00 In te ns ity 12 5. 71 12 6. 32 12 6. 38 12 7. 31 12 9. 22 13 0. 08 13 0. 12
4'-Methoxybiphenyl-4-carbaldehyde 13cc. 6 Yield 0.21 g (97 %).White crystalline powder, m.p.
106-107 oC (lit. m.p. 105-107 oC6). . 1H NMR (400 MHz, DMSO-d
6): δ= 3.80 (s, 3 H) 7.05 (d, J=8.8, 2H) 7.72 (d, J=8.8, 2H) 7.85 (d, J=8.2, 2H) 7.94 (d, J=8.2, 2H) 10.01 (s, 1 H). 13C NMR (100 MHz, DMSO-d6): δ = 55.2, 114.6, 126.6, 128.3, 130.2, 130.9, 134.4, 145.5, 159.8, 192.6. Calcd for C14H12O2: C 79.22; H 5.70. Found: C 79.16; H 5.78.
1H NMR of 13cc O C H3 O 10.0 9.5 9.0 8.5 8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 Chemical Shift (ppm) 3.05 2.03 2.00 1.00 DMSO-d6 M00 M04 M05 M03 M01 M02 2. 49 3. 80 7. 04 7. 06 7. 71 7. 73 7. 86 7. 93 7. 95 10 .0 1 8.00 7.75 7.50 7.25 7.00 Chemical Shift (ppm) 0 0.05 0.10 0.15 0.20 0.25 In te ns ity 2.03 2.01 2.02 2.00 M04 M03 M02 M01 7. 95 7. 93 7. 86 7. 84 7. 73 7.71 7.06 7.04 13C NMR of 13cc O C H3 O DMSO-d6 19 2. 57 9. 81 45 13 0. 15 12 8. 34 12 6. 61 11 4. 56 55 .2 3 39 .9 2 39 .7 1 39 .5 0 39 .2 8 39 .0 8 135 130 125 Chemical Shift (ppm) 0.25 0.50 In te ns ity 12 6. 61 12 8. 34 13 0. 15 13 0. 92 13 4. 45
4'-Formylbiphenyl-4-carboxylic acid 13dc. 7 Yield 0.21 g (93 %). White crystalline powder, m.p. 313-315 oC (decomp.) (m.p. 314 oC7); 1H NMR (400 MHz, DMSO-d
6): δ = 7.87 (d, J = 8.3, 2H) 7.95 (d, J = 8.1 Hz, 2H) 8.01 (d, J = 8.1 Hz, 2H) 8.05 (d, J = 8.3 Hz, 2H) 10.06 (s, 1 H). 13C-NMR (100 MHz, DMSO-d6): δ = 127.3, 127.7, 130.03, 130.2, 130.8, 135.6, 142.8, 144.7, 167.0, 192.8. Calcd for C14H10O3: C 74.33; H 4.46, Found: C 74.29; H 4.51. 1H NMR of 13dc O OH O 10.5 10.0 9.5 9.0 8.5 8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 Chemical Shift (ppm) 2.03 0.97 DMSO-d6 M04 M02 M03 M00 M01 2. 49 2. 49 7. 86 7. 88 7. 96 8. 00 8. 04 8. 06 10 .0 6 8.2 8.1 8.0 7.9 7.8 7.7 Chemical Shift (ppm) 0 0.25 0.50 0.75 1.00 In te ns ity 2.03 2.01 2.00 2.00 M02 M03 M00 M01 8. 06 8. 04 8. 02 8. 00 7. 96 7. 94 7. 88 7. 86 13C NMR of 13dc O OH O 190 180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 Chemical Shift (ppm) DMSO-d6 12 7. 31 12 7. 72 13 0. 03 13 0. 19 13 0. 82 13 5. 61 14 2. 78 14 4. 66 16 7. 05 19 2. 77 136 134 132 130 128 126 Chemical Shift (ppm) 0.25 0.50 0.75 1.00 In te ns ity 13 5. 61 13 0. 82 13 0. 19 13 0. 03 12 7. 72 12 7. 31
2'-Formylbiphenyl-4-carboxylic acid 13dd. 8 Yield 0.22 g (95 %). White crystalline powder, m.p. 218-220 oC (m.p. 215-219 oC8); 1H NMR (400 MHz, DMSO-d 6): δ= 7.53 (d, J = 7.7, 1H), 7.56 (d, J = 8.2, 2H), 7.62 (t, J = 7.6, 1H), 7.76 (t, J = 7.6, 1H), 7.94 (d, J = 7.7, 1H), 8.04 (d, J = 8.2, 2H), 9.87 (s, 1 H), 13.12 (br.s, 1H). 13C-NMR (100 MHz, DMSO-d 6): δ = 127.9, 128.5, 129.4 130.2, 130.4, 130.9, 133.2, 134.0, 141.8, 143.9, 167.1, 191.5. Calcd for C14H10O3: C 74.33; H 4.46, Found: C 74.25; H 4.54. 1H NMR of 13dd O O OH DMSO-d6 M03 M04 M02 M01 M05 M00 2. 49 7. 52 7. 54 7. 55 7. 57 7. 62 7. 63 78 7. 93 8. 03 8. 05 9. 87 8.2 8.1 8.0 7.9 7.8 7.7 7.6 7.5 7.4 Chemical Shift (ppm) 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1.0 In te ns ity 1.99 1.01 1.01 1.02 1.96 1.04 M03 M04 M02 M01 M00 8. 05 8. 03 7. 95 7.93 7. 78 7.78 7. 76 7.76 7. 75 7. 74 7.63 7. 62 7. 60 7. 57 7. 55 7. 54 7. 52
13C NMR of 13dd O O OH 190 180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 Chemical Shift (ppm) DMSO-d6 19 1. 52 16 7. 06 14 3. 88 14 1. 83 13 3. 99 13 0. 87 13 0. 22 12 9. 35 12 7. 86 136 134 132 130 128 126 Chemical Shift (ppm) 0.1 0.2 0.3 0.4 0.5 In te ns ity 12 7. 86 12 8. 54 12 9. 35 13 0. 22 13 0. 35 13 0. 87 13 3. 20 13 3. 99
4'-Trifluoromethylbiphenyl-4-carboxylic acid 13de.9 Yield 0.26 g (98 %). White crystalline powder,
m.p. 121-122 oC (m.p. 120.0-121.1oC9); 1H NMR (400 MHz, DMSO-d
6): δ= 7.86–7.82 (m, 4H), 7.94 (d, J = 8.2 Hz, 2H), 8.05 (d, J = 8.3 Hz, 2H), 13.05 (br s, 1H). 13C-NMR (100 MHz, DMSO-d
6): δ = 124.2 (q, JC-F = 270), 125.9 (m), 127.3, 127.8, 128.5 (q, JC-F = 32 Hz), 130.0, 130.6, 142.6, 143.0, 167.0. Calcd for C14H9F3O2: C, 63.16; H, 3.41, Found C, 63.11; H, 3.49.
1H NMR of 13de F F F O OH 9.0 8.5 8.0 7.5 Chemical Shift (ppm) 4.05 2.00 2.00 M02 M01 M00 7. 82 7. 84 7. 86 7. 93 7. 95 8. 04 8. 06 3.5 3.0 2.5 2.0 Chemical Shift (ppm) 0 0.25 0.50 0.75 1.00 In te ns ity DMSO-d6 2. 49 2. 49 14.5 14.0 13.5 13.0 12.5 12.0 Chemical Shift (ppm) 0 0.05 0.10 In te ns ity 0.99 M03 13 .0 5
13C NMR of 13de 170 160 150 140 130 120 110 100 90 80 70 60 50 40 Chemical Shift (ppm) 0 0.05 0.10 0.15 0.20 0.25 0.30 0.35 0.40 0.45 0.50 In te ns ity DMSO-d6 16 6. 99 14 3. 03 142. 64 13 0. 55 13 0. 04 12 7. 82 12 7. 27 12 5. 90 12 5. 60 12 2. 90 12 0. 15 39 .5 0 F F F O OH Region of 13C NMR of 13de 0.05 0.10 0.15 0.20 0.25 In te ns ity 13 0. 55 13 0. 04 01 12 8. 69 12 8. 37 12 7. 82 12 7. 27 12 5. 90 12 5. 86 5. 60 2. 90 F F F O OH
4-Methoxy-4'-nitrobiphenyl13cf. 10 Yield 0.22 g (97 %). Yellow crystalline powder, m.p. 110-111 °C (m.p. 109-110 °C10). 1H NMR (400 MHz, DMSO-d
6): δ = 3.81 (s, 3 H) 7.06 (d, J=8.62 Hz, 2H) 7.73 (d, J=8.62 Hz, 2H) 7.88 (d, J=8.63 Hz, 2H) 8.24 (d, J=8.63 Hz, 2H). 13C NMR (100 MHz, DMSO-d6): δ = 55.3, 114.7, 124.1, 127.0, 128.6, 129.9, 146.0, 146.3, 160.2.. Calcd for C13H11NO3: C 68.11; H 4.84, N 6.11. Found: C 68.06; H 4.89, N 6.08. 1H NMR of 13cf N+ O -O O CH3 8.5 8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 Chemical Shift (ppm) 3.02 2.02 2.00 1.99 DMSO-d6 M04 M01 M00 M02 M03 3. 81 7. 05 7. 07 7. 72 7. 74 7. 87 7. 90 8. 22 8. 25 8.5 8.0 7.5 7.0 Chemical Shift (ppm) 0 0.1 0.2 0.3 0.4 0.5 In te ns ity 2.02 2.01 2.00 1.99 M05 J(M01)=8.6 Hz M04 M01 M02 M03 8. 25 8. 22 7. 90 7. 87 7.74 7.72 7.07 7. 05 13C NMR of 13cf N+ O -O O CH3 160 152 144 136 128 120 112 104 96 88 80 72 64 56 48 40 32 Chemical Shift (ppm) DMSO-d6 38 .8 8 39 .0 8 39 .2 9 39 .5 0 39 .7 1 39 .9 2 40 .1 3 55 .3 0 11 4. 66 12 4. 07 12 6. 96 12 8. 56 12 9. 91 14 5. 97 14 6. 26 16 0. 18 160 152 144 136 128 120 112 Chemical Shift (ppm) 0.1 0.2 0.3 0.4 0.5 In te ns ity 16 0. 18 14 6. 26 14 5. 97 12 9. 91 12 8. 56 12 6. 96 12 4. 07 11 4. 66
4-Nitrophenylboronic acid 12f.* From Sigma-Aldrich. Yellow crystalline powder. 1H NMR (400 MHz, DMSO-d6): δ= 8.01 (d, J = 8.6 Hz, 2H) 8.16 (d, J = 8.6 Hz, 2H), 8.47 (br.s, 2H). 13C NMR (100 MHz, DMSO-d6): δ= 122.1, 135.2, 142.4, 148.7. 1H NMR of 12f B OH O H N+ O -O 9.0 8.5 8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 Chemical Shift (ppm) 2.01 DMSO-d6 M01 M00 8. 00 8. 02 8. 15 8. 17 8. 47 9.0 8.5 8.0 7.5 Chemical Shift (ppm) 0 0.25 0.50 0.75 1.00 In te ns ity 2.01 2.00 M01 M00 8. 47 8. 17 8. 15 8. 02 8. 00 13C NMR of 12f B OH O H N+ O -O DMSO-d6 39 .0 8 39 .2 9 39 .5 0 39 .7 1 39 .9 2 12 2. 07 13 5. 25 0.1 0.2 0.3 0.4 0.5 In te ns ity 14 8. 74 2. 44 13 5. 25 12 2. 07
Gram-scale synthesis of 2-amino-4′-chlorobiphenyl 16.
A 250 mL flask with a magnetic stir bar was charged with 2-bromoaniline 14 (1.72 g, 10 mmol), 4-chlorophenylboronic acid 15 (1.72 g, 11 mmol), K2CO3 (3.45 g, 25 mmol), Bu4NBr (0.0322 g, 0.1 mmol, 1 mol%), 100 ml of H2O and 100 μL of solution (510-3 M) of palladium complexes Pd2Cl2(L2 -H)2 (0.01 mol%) in MeOH under air atmosphere. The reaction mixture was placed in an oil bath preheated to 160 °C and stirred under reflux for 10 min. After this time, the mixture was diluted with 100 mL of H2O and stirred under reflux for additional 30 min, then, cooled to room temperature, filtered and the filter cake was further crystallized from hexane giving 1.976 g (97%) product 16. This method avoids the use of a large amount of organic solvents during separation process, which would be more favorable from a safe and environmental perspective.
4'-Chlorobiphenyl-2-amine 16.11 White powder, m.p. 48-49 oC (m.p. 47-48 oC11). 1H NMR (400
MHz, CDCl3): δ = 3.63 (brs, 2H, NH2), 6.78 (dd, 1H, J = 7.9, J = 1.0,), 6.85 (td, 1H, J = 7.6, 1.0,), 7.11 (dd, 1H, J = 7.6, J = 1.5), 7.19 (td, J = 7.9, 1.0, 1H), 7.38-7.46 (m, 4H). 13C NMR (100 MHz,, CDCl3): δ= 115.7, 118.7, 126.2, 128.8, 128.9, 130.3, 130.4, 133.0, 137.8, 143.4. Mass spectrum, m/z (I, %): 205 [37Cl-M]+ (27), 204 (12), 203 [35Cl-M]+ (100), 202 (13), 169 (17), 168 (55), 167 (34), 166 (16), 83 (26). 1H NMR of 16
13C NMR of 16
Gram-scale synthesis of2',4'-difluoro-4-hydroxy-[1,1'-biphenyl]-3-carboxylic acid (diflunisal)
19.
A 250 mL flask with a magnetic stir bar was charged with 5-iodosalicylic acid 17 (2.64 g, 10 mmol), 2,4-difluorohenylboronic acid 18 (1.74 g, 11 mmol), K2CO3 (3.45 g, 25 mmol), 100 ml of H2O and 100 μL of solution (510-5 M) of palladium complexes Pd
2Cl2(L2-H)2 (0.0001 mol%) in MeOH under air atmosphere. The reaction mixture was placed in an oil bath preheated to 160 °C and stirred under reflux for 5 min. After this time, the mixture was cooled, diluted with 50 mL of H2O, 50 mL of Et2O and acidified by 1 M HCl. The organic phase was separated, and the aqueous layer was extracted with Et2O (2 x 25 mL). The combined ether layers were washed with brine (2 x 50 mL), dried over Na2SO4 and filtered through silica gel pad. Evaporation of ether gave a crude material, which was recrystallized from water-methanol mixture (9:1) to give 2.45 g (98%) product 19.
1H NMR of 19
General procedure for the Mizoroki–Heck reaction
A 20 mL Schlenk tube with a magnetic stir bar was charged with 3-bromobenzoic acid 11 (0.5 mmol), acrylic acid 20 (0.75 mmol), K2CO3 (1.5 mmol), 5 ml of water and 50 μL of the solution (510 -3 M) of palladium complexes Pd
2Cl2(L-H)2 in MeOH under air atmosphere. The reaction mixture was placed in a preheated at 160 °C oil bath and stirred for the appropriate time (Tables 4S). After this time, the mixture was cooled, diluted with 5 mL of H2O, 5 mL of EtOAc and acidified by 1 M HCl. The organic phase was separated, and the aqueous layer was extracted with EtOAc (2 x 5 mL). The combined organic layers were washed with brine (5 mL) and dried over Na2SO4. The yield was determined by 1H NMR analysis with 1,1,2,2-tetrachloroethane (0.5 mmol) as internal standard (Tables 4S). The pure product was obtained by a simple filtration of solution through silica gel pad and evaporation of solvent.
3-[(E)-2-carboxyvinyl]benzoic acid 21.13 White crystalline powder (0.092 g, 96 % yield), m.p. 284–
285 °C (m.p. 284–286 °C13). 1H NMR (400 MHz, DMSO-d
6): δ= 6.58 (d, 1H, J = 16.0), 7.53 (t, 1H, J = 7.8), 7.64 (d, 1H, J = 16.0), 7.90-8.00 (m, 2H), 8.14 (s, 1H), 12.81 (br. s, 2H). 13C NMR (100 MHz, DMSO-d6): δ = 120.5, 129.0, 129.3, 130.8, 131.6, 132.0, 134.7, 142.9, 166.9, 167.4. Calcd for C10H8O4: C 62.50; H 4.20; O 33.30. Found: C 62.44; H 4.28.
Table 4S. Mizoroki–Heck reaction of m-bromobenzoic acid 11 and acrylic acid 20 in the presence of complexes Pd2Cl2(L-H)2a
Entry Pd2Cl2(L-H)2 Time, min Yield,b %
1 Pd2Cl2(L1-H)2 20 97 2 Pd2Cl2(L2-H)2 20 100 (96)c 3 Pd2Cl2(L2-H)2 200 (20 °C) 0 4 Pd2Cl2(L3-H)2 20 95 5 Pd2Cl2(L4-H)2 20 98 6 Pd2Cl2(L4-H)2 200 (20 °C) 0
a Aryl halide (0.5 mmol), acrylic acid (0.6 mmol), K
2CO3 (1.25 mmol), 5 mL of H2O, 100 °С.
b1H NMR yield with 1,1,2,2-tetrachloroethane (0.5 mmol) as internal standard. c Isolated yield. 1H NMR of 21 13.0 12.5 12.0 11.5 11.0 10.5 10.0 9.5 9.0 8.5 8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 Chemical Shift (ppm) 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1.0 In te ns ity 2.001.00 1.01 DMSO-d6 M01 M02 M04 M00 M03 2. 49 2. 49 6. 56 6. 60 7. 51 7. 53 7. 62 7. 66 7. 94 7. 96 8. 14 12 .8 1 O OH O OH 21 8.0 7.5 7.0 6.5 Chemical Shift (ppm) 0.50 0.75 1.00 In te ns ity 2.00 1.01 1.01 1.00 0.98 M01 M02 M04 M00 M03 8. 14 7.96 7. 94 7. 94 7. 66 7.62 7. 55 7. 53 7. 51 6. 60 6. 56 13C NMR of 21 0.35 0.40 0.45 0.50 ns ity DMSO-d6 39 .0 9 39 .3 0 39 .5 0 39 .7 1 39 .9 2 O OH O OH 21 0.10 0.15 0.20 0.25 In te ns ity 134. 69 13 1. 99 13 1. 57 13 0. 75 129. 28 12 8. 99
Gram-scale synthesis of 2′-ethylhexyl (2E)-3-(4-methoxyphenyl)acrylate (octinoxate) 25.
A 250 mL flask with a magnetic stir bar was charged with 4-iodoanisole 26 (2.34 g, 10 mmol),
2′‐ethylhexyl acrylate 27 (11 mmol) (2.02 g, 11 mmol), K2CO3 (2.76 g, 20 mmol), Bu4NBr (0.0322 g, 0.1 mmol, 1 mol%), 100 ml of H2O and 100 μL of solution (510-3 M) of palladium complexes Pd2Cl2(L2-H)2 (0.01 mol%) in MeOH under air atmosphere. The reaction mixture was placed in an oil bath preheated to 160 °C and stirred under reflux for 25 min. After this time, the mixture was cooled and extracted with Et2O (3 x 25 mL). The combined ether layers were washed with brine (2 x 50 mL), dried over Na2SO4 and filtered through silica gel pad. Evaporation of ether under reduced pressure gave a crude material, low-temperature crystallization of which from hexane afforded 2.82 g (97%) of octinoxate 25.
2′-Ethylhexyl (2E)-3-(4-methoxyphenyl)acrylate (octinoxate) 25.14 Colorless oil. 1H NMR (400 MHz, CDCl3): δ = 0.93–0.97 (m, 6H), 1.34–1.45 (m, 8H), 1.64–1.74 (m, 1H), 3.85 (s, 3H), 4.09–4.12 (m, 2H), 6.34 (d, 1 H, J = 15.9), 6.92 (d, 2H, J = 8.7,), 7.50 (d, 2H, J = 8.7), 7.65 (d, 1H, J = 16.1,). 13C NMR (400 MHz, CDCl3):δ = 11.1, 14.1, 23.0, 23.9, 29.0, 30.5, 38.9, 55.4, 66.8, 114.3, 115.9, 127.2, 129.7, 144.1, 161.3, 167.5. Mass spectrum, m/z (I, %): 290 [M]+ (9), 179 (12), 178 (100), 161 (57), 134 (14), 133 (17).
13C NMR of 25
General procedure for the Sonogashira reaction
A 20 mL Schlenk tube with a magnetic stir bar was charged with methyl p-iodobenzoate 22 (0.5 mmol), prop-2-yn-1-ol 23 (0.65 mmol), K2CO3 (1.25 mmol), Bu4NBr (1 mol%), 5 ml of water and 50
μL of the solution (510-3 M) of palladium complexes Pd
2Cl2(L-H)2 in MeOH under air atmosphere. The reaction mixture was placed in a preheated at 160 °C oil bath and stirred for the appropriate time (Tables 5S). After this time, the mixture was cooled, diluted with 5 mL of H2O and 5 mL of EtOAc. The organic phase was separated, and the aqueous layer was extracted with EtOAc (2 x 5 mL). The combined organic layers were washed with brine (5 mL) and dried over Na2SO4. The yield was determined by 1H NMR analysis with 1,1,2,2-tetrachloroethane (0.5 mmol) as internal standard (Tables 5S). The pure product was obtained by a simple filtration of solution through silica gel pad and evaporation of solvent. Removal of solvent was carried out in a closed volume, which allowed carrying out 90-95% regeneration of the solvent.
Table 5S. Sonogashira reaction between methyl p-iodobenzoate 22 and prop-2-yn-1-ol 23 in the presence of complexes Pd2Cl2(L-H)2a
Entry Pd2Cl2(L-H)2 Time, min Yield,b %
1 Pd2Cl2(L1-H)2 10 95 2 Pd2Cl2(L2-H)2 10 100 (95)c 3 Pd2Cl2(L2-H)2 150 (20 °C) 98 4 Pd2Cl2(L3-H)2 10 93 5 Pd2Cl2(L4-H)2 10 96 6 Pd2Cl2(L4-H)2 200 (20 °C) 87
a Aryl halide (0.5 mmol), prop-2-yn-1-ol (0.65 mmol), K
2CO3 (1.25 mmol), Bu4NBr (1 mol%), °С.
b1H NMR yield with 1,1,2,2-tetrachloroethane (0.5 mmol) as internal standard. c Isolated e yield. 1H NMR of 24 8.5 8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 Chemical Shift (ppm) 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1.0 In te ns ity 3.11 2.04 2.02 2.00 Chloroform-d M00 M03 M01 M02 7. 97 7.95 7.47 7.45 4. 50 3. 90 8.00 7.75 7.50 7.25 Chemical Shift (ppm) 0 0.1 0.2 In te ns ity 2.02 2.00 Chloroform-d M03 M02 7. 45 7. 47 7. 95 7. 97 O OMe OH 24 2.5 2.0 1.5 Chemical Shift (ppm) 0 0.25 0.50 0.75 1.00 In te ns ity 0.99 2. 00 13C NMR of 24 170 165 160 155 150 145 140 135 130 125 120 115 110 105 100 95 90 85 80 75 70 65 60 55 50 45 Chemical Shift (ppm) 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1.0 In te ns ity Chloroform-d 51 .5 3 52 .2 5 76 .6 9 77 .0 0 77 .3 2 84 .8 6 90 .2 1 12 7. 23 12 9. 46 12 9. 74 13 1. 56 16 6. 52 O OMe OH 24 132 130 128 126 124 122 Chemical Shift (ppm) 0.25 0.50 0.75 1.00 In te ns ity 13 1. 56 12 9. 74 12 9. 46 12 7. 23
Gram-scale synthesis of2-(phenylethynyl)aniline 28.
A 250 mL flask with a magnetic stir bar was charged with 2-iodoaniline 29 (2.19 g, 10 mmol), phenylacetylene 30 (1.13 g, 11 mmol), K2CO3 (2.76 g, 20 mmol), Bu4NBr (0.0322 g, 0.1 mmol, 1 mol%), 100 ml of H2O and 100 μL of solution (510-3 M) of palladium complexes Pd2Cl2(L2-H)2 (0.01 mol%) in MeOH under air atmosphere. The reaction mixture was placed in an oil bath preheated to 160 °C and stirred under reflux for 20 min. After this time, the reaction mixture was cooled and extracted with Et2O (3 x 25 mL). The combined ether layers were washed with brine (2 x 50 mL), dried over Na2SO4 and filtered through silica gel pad. After evaporation of the ether under reduced pressure, the resulting crude material was recrystallized from hexane to obtain 1.84 g (95%) of 2- (phenylethynyl)aniline. 28.
2-(Phenylethynyl)aniline 28.16 Colourless powder, mp 91–92 °C (mp 91–92 °C16). 1H NMR (400
MHz, CDCl3): δ = 4.74 (brs, 2H), 6.77 (t, 2H, J = 8.5), 7.14 (m, 1H), 7.34 (m, 4H), 7.54 (m, 2H). 13C NMR (100 MHz, CDCl3): δ = 86.0, 94.8, 108.1, 114.5, 118.1, 123.4, 128.3, 128.5, 129.8, 131.6, 132.3, 147.9. IR (KBr): 3434, 3311, 3054, 1463, 1257, 1158, 988 cm-1. Mass spectrum, m/z (I, %): 194 [M+H]+ (16), 193 [M]+ (100), 192 (17), 165 (28).
13C NMR of 28
Mass-spectra of complexes Pd2Cl2(L-H)2 (L=L2, L4)
Area of mass-spectrum of complex Pd2Cl2(L2-H)2: isotopic peaks for ion [M – 2Cl+2CH3CN]2+
Area of predicted mass-spectrum of complex Pd2Cl2(L2-H)2: isotopic peaks for ion [M –
Area of mass-spectrum of complex Pd2Cl2(L2-H)2: isotopic peaks for ion [M –2Cl+CH3CN+CHO2]+
Area of predicted mass-spectrum of complex Pd2Cl2(L2-H)2: isotopic peaks for ion [M –
Mass-spectrum of complex Pd2Cl2(L4-H)2
Area of predicted mass-spectrum of complex Pd2Cl2(L4-H)2: isotopic peaks for ion [M –
2Cl+2CH3CN]2+
Area of predicted mass-spectrum of complex Pd2Cl2(L4-H)2: isotopic peaks for ion [M –
2Cl+CH3CN+CHO2]+
Comparative results for the synthesis of biaryls by Suzuki reaction using reported catalytic systems versus the present method
Corresponding results are presented in Table 6S. While reported TON and TOF of the best proposed here catalyst are comparable with our previous results (entry 7), the ligand framework presented here is much easier to prepare.
Table 6S. Comparative results for the synthesis of biaryls by Suzuki reaction using reported catalytic systems versus the present method
Entry Pd source Ligand Mol% Conditions Yield,
% TON/TOF, h-1 Ref. 1 (AllPdCl)2 Tedicyp 1x10-6 K2CO3, xylene, 130 °C, 20 h, 97 9.7x107/5x105 17
4 PdCl2 Imino-pyridine 10−4 K2CO3, toluene, 110 °C, 48 h, N2 99 9.9×105/2x104 20 5 Pd(COD)Cl2 PCN-pincer 10−5 K2CO3, H2O, 100 °C 82 8.2×106/- 21 6 PdCl2 N-Heterocyclic carbene (NHC) 10−4 K 2CO3, DMF-H2O, 110 °C, 2 h, N2 100 1×106/5×105 22 7 PdCl2 1,2,3-triazol-isoxazole 10−4 K 2CO3, H2O, 100 °C, 5 min 100 1×106/1.2×107 23 8 PdCl2 N-(pyrimidin- 2-yl)-1,2- azole-3-carboxamide 10−4 K 2CO3, H2O, 100 °C, 5 min 100 1×106/1.2×107 This work Computational studies
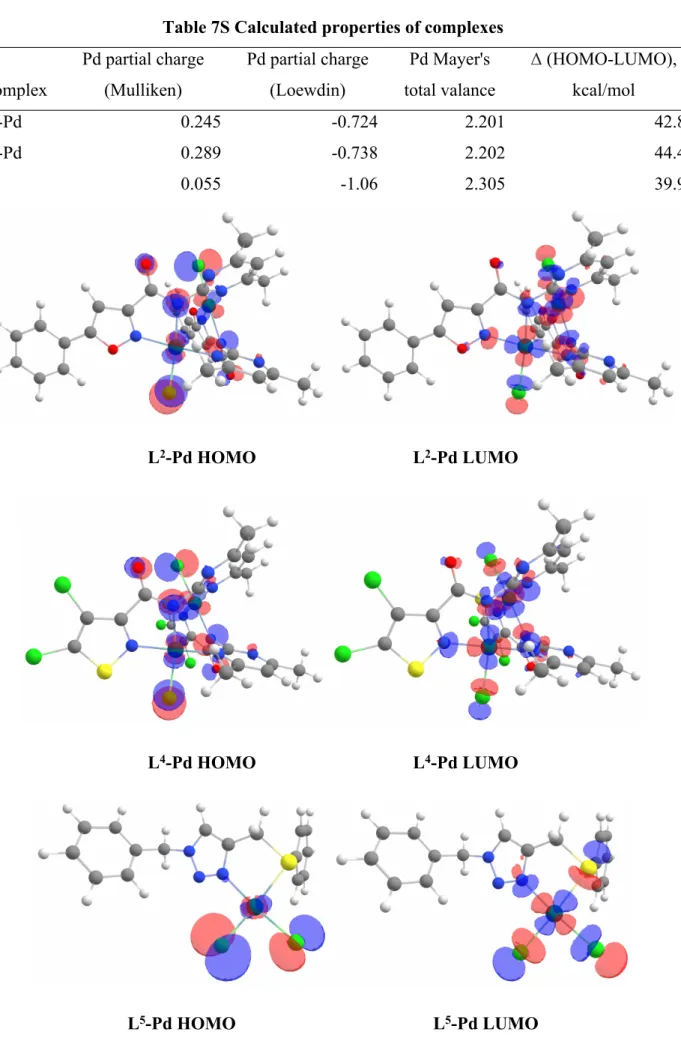
Quantum-chemical calculations were performed to somehow rationalize the observed high catalytic activity, as well as the absence of palladium black. Computational studies were also performed for the triazole complex L5 [24], which was chosen as a random referential catalytically active azolic complex. Geometrical parameters obtained from single-crystal X-Ray analysis of corresponding metal complexes were used as a starting point for further optimization of structures at the PW91-LDA/SV(P) level of theory with LANLTZ basis set and corresponding ECP for Pd atoms. The calculations were performed using ORCA software [25], the visualization of frontiers orbitals was accomplished using ChemCraft [26]. The global minimum for the obtained complexes was proved by the absence of imaginary frequencies for optimized geometries of complexes. Some of the obtained parameters are provided in table 7S; visualized frontier orbitals are presented in figure 2.
Table 7S Calculated properties of complexes Complex Pd partial charge (Mulliken) Pd partial charge (Loewdin) Pd Mayer's total valance Δ (HOMO-LUMO), kcal/mol L2-Pd 0.245 -0.724 2.201 42.8 L4-Pd 0.289 -0.738 2.202 44.4 L5 0.055 -1.06 2.305 39.9 L2-Pd HOMO L2-Pd LUMO L4-Pd HOMO L4-Pd LUMO
The difference in the studied complexes activity within 1,2-azolic row might be partially explained by the lower HOMO-LUMO gap (42.8 kcal/mol for L2-Pd and 44.4 kcal/mol for L4-Pd), as well as
lower partial charge in the case of isoxazole complex. However, that could not explain the higher activity compared to the REF complex, as well as the absence of palladium black. The obtained results could be partially rationalized by the differences in the reaction conditions, but we assume that it might go beyond that. The detailed studies on this topic will be provided in follow-up publications.
References
1. M. C. Burla, R. Caliandro, B. Carrozzini, G. L. Cascarano, C. Cuocci, C. Giacovazzo, M. Mallamo, A. Mazzone, G.
Polidori, Crystal structure determination and refinementvia SIR2014. J. Appl. Cryst., 2015, 48, 306.
2. G. M. Sheldrick, Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C: Cryst. Struct. Commun.,
2015, 71, 3.
3. A. L. Spek, Structure validation in chemical crystallography. Acta Crystallogr. Sect. D: Biol. Crystallogr., 2009, 65,
148.
4. D. V. Evdokimov, N. A. Bumagin, A PdCl2 complex with isonicotinaldehyde oxime, an efficient catalyst for the
Suzuki reaction in aqueous media. Russ. Chem. Bull. Int. Ed., 2007, 56, 369.
5. F. Picard, T. Schulz, R. W. Hartmann, 5-Phenyl substituted 1-methyl-2-pyridones and 4′-substituted
biphenyl-4-carboxylic acids. synthesis and evaluation as inhibitors of steroid-5α-reductase type 1 and 2, Bioorg. Med. Chem., 2002, 10,
437.
6. L. Huang, L. K. G. Ackerman, K. Kang, A. M, Parsons, D. J. Weix, LiCl-Accelerated multimetallic cross-coupling of
aryl chlorides with aryl triflates, J. Amer. Chem. Soc., 2019, 141, 10978.
7. Benzoxazolyl-phenyl-stilbene, Patent Ciba-Geigy AG, DE2848193, 1979.
8. Pyrrolobenzodiazepine pyridine carboxamides and derivatives as follicle-stimulating hormone receptor antagonists,
Patent Wyeth, US2006287522, 2006.
9. R. Berini, S. Cacchi, G.Fabrizi, G. Forte, F. Petrucci, A. Prastaro, S. Niembro, A. Shafir, A. Vallribera,
Perfluoro-tagged, phosphine-free palladium nanoparticles supported on silica gel: application to alkynylation of aryl halides, Suzuki–
Miyaura cross-coupling, and Heck reactions under aerobic conditions, Green Chem. 2010, 12, 150.
10. K. V. Kutonova, N. Jung, M. E. Trusova, V. D. Filimonov, P. S. Postnikov, S. Bräse, Arenediazonium Tosylates
(ADTs) as Efficient Reagents for Suzuki–Miyaura Cross-Coupling in Neat Water, Synthesis, 2017, 49, 1680.
11. E. H. Huntress, M. K. Seikel, Fluorenones and Diphenic Acids. VI. Ring Cleavage of Chloro-, Hydroxy-,
2-Amino- and 2-Sulfofluorenones with Potassium Hydroxide in Diphenyl Ether. J. Am. Chem. Soc.,1939, 61, 816.
12. J. Hannah, W. V. Ruyle, H. Jones, A. R. Matzuk, K. W. Kelly, B. E. Witzel, W. J. Holtz, R. A. Houser, and T. Y.
Shen, Novel analgesic-antiinflammatory salicylates. J. Med. Chem., 1978, 21, 1093.
13. D. V. Evdokimov, N. A. Bumagin, 4-Dimethylaminopyridine, an efficient ligand for the Heck reaction in aqueous
media. Russ. Chem. Bull., 2007, 56, 1093.
14. J. G. De Vries, The Heck reaction in the production of fine chemicals. Can. J. Chem., 2001, 79, 1086.
15. N. Asano, K. Sasaki, I. Chataigner, M. Shigeno, Y. Kondo, Sodium Phenoxide Mediated Hydroxymethylation of
Alkynylsilanes with N ‐[(Trimethylsiloxy)methyl]phthalimide . European J Org Chem., 2017, 6926.
16. Y. Yin, W. Ma, Z. Chai, G. Zhao, Et2Zn-Catalyzed Intramolecular Hydroamination of Alkynyl Sulfonamides and the
Related Tandem Cyclization/Addition Reaction. J. Org. Chem., 2007, 72, 5731.
17. M. Feuerstein, D. Laurenti, H. Doucet, M. Santelli, Palladium–Tetraphosphine Complex: An Efficient Catalyst for
18. G. Zhang, Y. Luan, X. Han, Y. Wang, X. Wen, C. Ding, J. Gao, A palladium complex with functionalized
β-cyclodextrin: a promising catalyst featuring recognition abilities for Suzuki–Miyaura coupling reactions in water, Green
Chem, 2013, 15, 2081.
19. Q. Yang, L. Wang, L. Lei, X.-L. Zheng, H.-Y. Fu, M.-L. Yuan, H. Chen, R.-X. Li, PdCl2
-2,6-bis(1,5-diphenyl-1H-pyrazol-3-yl)pyridine catalyzed Suzuki–Miyaura cross-coupling, Catal. Commun., 2012, 29, 194.
20. Y. Tang, Y. Zeng, Q. Hu, F. Huang, L. Jin, W. Mo, N. Sun, B. Hu, Z. Shen, X. Hu, W.-H. Sun, Efficient Catalyst for Both Suzuki and Heck Cross-Coupling Reactions Synthesis and Catalytic Behaviour of Geometry-Constrained
Iminopyridylpalladium Chlorides, Adv. Synth. Catal., 2016, 358, 2642.
21. B. Ines, R. SanMartin, F. Churruca, E. Domínguez, M.K. Urtiaga, M.I. Arriortua, A Nonsymmetric Pincer-Type
Palladium Catalyst In Suzuki, Sonogashira, and Hiyama Couplings in Neat Water, Organometallics, 2008, 27, 2833.
22. W. Huang, J. Guo,Y. Xiao, M. Zhu, G. Zou, J. Tang, Palladium–benzimidazolium salt catalyst systems for Suzuki coupling: development of a practical and highly active palladium catalyst system for coupling of aromatic halides with
arylboronic acids, Tetrahedron, 2005, 61, 9783.
23. N. A Bumagin, A. V. Kletskov, S. K. Petkevich, I.A. Kolesnik, A.S. Lyakhov, L. S. Ivashkevich, A. V. Baranovsky,
P. V. Kurman, V. I. Potkin, Substituted 1-(isoxazol-3-yl)methyl-1H-1,2,3-triazoles: Synthesis, palladium(II) complexes,
and high-turnover catalysis in aqueous media, Tetrahedron, 2018, 74, 3578.
24. F. Saleem, G. K. Rao, A. Kumar, S. Kumar, M. P. Singh, A. K. Singh, Palladium(ii) Complexes Bearing the 1,2,3-Triazole Based Organosulfur/ Selenium Ligand: Synthesis, Structure and Applications in Heck and Suzuki–Miyaura
Coupling as a Catalyst via Palladium Nanoparticles. RSC Adv. 2014, 4, 56102.
25. F. Neese, The ORCA Program System. WIREs Comput. Mol. Sci. 2012, 2 , 73.