Minocycline Hepatotoxicity: Clinical characterization and
identification of HLA-B* 35:02 as a risk factor
Thomas Jacob Urban1,2, Paola Nicoletti3, Naga Chalasani4, Jose Serrano5, Andrew Stolz6, Ann K. Daly7, Guruprasad P. Aithal8, John Dillon9, Victor Navarro10, Joseph Odin11, Huiman Barnhart12, David Ostrov13, Nanye Long1,2, Elizabeth Theresa Cirulli12, Paul Brent Watkins1,2, and Robert John Fontana14 on behalf of the Drug-Induced Liver Injury Network (DILIN), the Pharmacogenetics of Drug-Induced Liver Injury group (DILIGEN) and the International Serious Adverse Events Consortium (iSAEC)
1Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC, USA 2UNC Institute for Drug Safety Sciences, Durham, NC, USA
3Center for Computational Biology and Bioinformatics, Columbia University, New York, NY, USA 4Indiana University School of Medicine, Indianapolis, IN, USA
5National Institute of Diabetes Digestive and Kidney Diseases, Bethesda, MD, USA 6University of Southern California, Los Angeles, CA, USA
7Newcastle University, Newcastle upon Tyne, United Kingdom
8National Institute for Health Research Nottingham Digestive Diseases Biomedical Research Unit,
Nottingham University Hospitals NHS Trust and University of Nottingham, United Kingdom
9University of Dundee, Dundee, United Kingdom
To whom correspondence should be addressed: Robert J. Fontana, MD, Professor of Medicine, 3912 Taubman Center, Ann Arbor, MI 48109-0362, Tel: (734)-936-4780, Fax: (734)-936-7392, [email protected].
Author e-mail addresses: Guruprasad P Aithal [email protected] Huiman Barnhart [email protected]
Naga Chalasani [email protected] Elizabeth T. Cirulli [email protected] Ann Daly [email protected] John Dillon [email protected] Robert Fontana [email protected] David Kleiner [email protected] Nanye Long [email protected] Victor Navarro [email protected] Paola Nicoletti [email protected] Joseph Odin [email protected] David Ostrov [email protected]
Jose Serrano [email protected] Andrew Stolz [email protected]
Thomas Urban [email protected] Paul Watkins [email protected]
Specific Author contributions: All of the authors were involved in study concept and design, acquisition of data, analysis and interpretation of data, and critical review of the final draft of the manuscript. Drafting of the manuscript (Fontana, Urban, Watkins, Aithal, Barnhart, Kleiner, Ostrov), statistical analyses (Urban, Long, Nicoletti, Barnhart), review and finalization (all listed authors) Potential Conflicts of interest: Dr's Barnhart, Kleiner, Stolz, Urban, Long, Aithal, Daly, Dillon, Cirulli and Serrano have no conflicts
HHS Public Access
Author manuscript
J Hepatol
. Author manuscript; available in PMC 2017 October 10.Published in final edited form as:
J Hepatol. 2017 July ; 67(1): 137–144. doi:10.1016/j.jhep.2017.03.010.
A
uthor Man
uscr
ipt
A
uthor Man
uscr
ipt
A
uthor Man
uscr
ipt
A
uthor Man
uscr
10Einstein Medical Center, Philadelphia, PA, USA
11Icahn School of Medicine at Mount Sinai, New York, NY, USA 12Duke University, Durham, NC, USA
13University of Florida College of Medicine, Gainesville, FL, USA 14University of Michigan, Ann Arbor, MI, USA
Abstract
Background & Aims—Minocycline hepatotoxicity can present with prominent autoimmune features in previously healthy individuals. The aim of this study was to identify genetic determinants of minocycline DILI in a well-phenotyped cohort of patients.
Methods—Caucasian patients with minocycline DILI underwent genome-wide genotyping and were compared to unexposed population controls. Human leukocyte antigen (HLA) binding of minocycline was assessed using AutoDock Vina.
Results—Amongst the 25 cases, 80% were female, median age was 19 years and median latency from drug start to DILI onset was 318 days. At presentation, 76% had acute hepatocellular liver injury, median ALT 1077 U/L (range: 63 to 2333), median bilirubin 4.5 mg/dl (range: 0.2 to 16.7), and 90% had a +ANA. During follow-up, 50% were treated with corticosteroids and no subjects died or required a liver transplant. A significant association was noted between HLA-B*35:02 and risk for minocycline-DILI; a 16% carrier frequency in DILI cases compared to 0.6% in population controls (Odds Ratio: 29.6, 95% CI: 7.8-89.8, p=2.5 × 10-8). Verification of HLA-B*35:02 imputation was confirmed by sequence-based HLA typing. HLA-B*35:02 carriers had similar presenting features and outcomes compared to non-carriers. In silico modeling studies support the hypothesis that direct binding of minocycline to this novel HLA risk allele might be an important initiating event in minocycline-DILI.
Conclusion—HLA-B*35:02 is a rare HLA allele that was more frequently identified in the 25 minocycline-DILI cases compared to population controls. If confirmed in other cohorts, this HLA allele may prove to be a useful diagnostic marker of minocycline DILI.
Keywords
Drug induced liver injury; single nucleotide polymorphism; genetic association; autoimmunity; human leukocyte antigen
Idiosyncratic drug induced liver injury (DILI) is an important cause of acute and chronic liver injury in western patients. In addition to being a leading reason for regulatory actions amongst drugs in development and in the marketplace, DILI also accounts for 13% of adults with acute liver failure (1). A recent population based study indicated that the annual incidence of DILI was 19.1 per 100,000 person years in Iceland and that antibiotics were the most commonly implicated agents (2). Similarly, analyses from the ongoing Drug induced Liver Injury Network (DILIN) prospective study in the United States also identified antibiotics as the leading cause of DILI, with amoxicillin-clavulanate being most frequently implicated (3). Prior reports have also implicated minocycline as a cause of DILI with
A
uthor Man
uscr
ipt
A
uthor Man
uscr
ipt
A
uthor Man
uscr
ipt
A
uthor Man
uscr
characteristic clinical features including systemic arthralgias and detectable autoantibodies arising in young women (4). Recent studies have suggested that various laboratory,
histological and clinical features can help differentiate auto-immune like DILI from sporadic autoimmune hepatitis but confirmatory studies are needed (4).
Several groups have begun studies to better define the presenting features, risk factors, and outcomes of patients with DILI. In addition to identifying improved causality assessment methods and DILI biomarkers, studies exploring the potential genetic susceptibility in these rare patients with DILI have been undertaken. Prior genome wide association studies (GWAS) have identified single nucleotide polymorphisms in the Human Leukocyte Antigen (HLA) locus that are associated with DILI susceptibility to several drugs (5-8). The aim of the current study is to report upon the presenting clinical features and outcomes of patients with DILI attributed to minocycline that have enrolled into the ongoing DILIN prospective and retrospective studies. In addition to exploring clinical phenotypes, we also set out to identify potential genetic susceptibility factors in patients with minocycline DILI compared to population controls, using GWAS and confirmatory sequence-based HLA typing. Finally, preliminary results exploring the potential mechanism of the HLA-B*35:02 association with minocycline DILI using in silico modelling are presented.
Methods
DILIN Prospective study
Most of the subjects were enrolled in the DILIN prospective study protocol. DILI onset was defined as the first date after a subject taking minocycline met the predefined laboratory criteria for study entry, of either a serum aspartate aminotransferase (AST) or alanine aminotransferase (ALT) level that exceeded 5 × the upper limit of normal (ULN) (or 5 × pretreatment baseline if baseline abnormal), a serum alkaline phosphatase (Alk P) that exceeded 2 × the ULN (or 2 × pretreatment baseline if baseline abnormal), a total bilirubin > 2.5 mg/dl, or an international normalized ratio (INR) greater than 1.5 on two consecutive blood draws. All study participants were enrolled within 6 months of DILI onset.
A detailed medical history was obtained at the baseline study visit and additional laboratory and radiological testing were performed to more fully characterize the DILI event and exclude competing etiologies. Specifically, testing for hepatitis A, B, C, HIV, autoantibodies including anti-nuclear antibody titers, CMV, and EBV infection were obtained at the local laboratory. Enrolled patients were seen for a follow-up study visit at 6 months after initial enrollment and those with evidence of chronic DILI within 6 months of DILI onset were asked to return for additional follow-up visits at 12 and 24 months (9). Chronic DILI was defined as having a persistently elevated serum AST, ALT, or alk phos level, histological evidence of liver injury, or clinical evidence of portal hypertension at 6 months after DILI onset. Written informed consent was obtained from subjects and the study was approved by the Institutional Review Boards of all participating clinical sites.
The severity of the DILI episode was categorized on a 5-point scale from mild (1), moderate (2), moderate-hospitalized (3), severe (4), and fatal (5), where a fatal score was assigned only if the patient died or had liver transplant due to DILI (9). Of note, the clinical features
A
uthor Man
uscr
ipt
A
uthor Man
uscr
ipt
A
uthor Man
uscr
ipt
A
uthor Man
uscr
of some of the minocycline patients were previously presented in a separate report (10). In addition, clinical features from 2 of these cases have been posted as brief vignettes on the LiverTox website (see http://livertox.nlm.nih.gov/minocycline).
DILIN Retrospective study
DNA samples and phenotypic data from subjects with minocycline hepatotoxicity enrolled in the DILIN retrospective study were included. Study inclusion criteria were patients that developed DILI due to one of 8 prespecified drugs that included minocycline with a DILI onset date after 1994. Subjects had to have a total bilirubin of > 2.5 mg/dl and a complete set of labs at DILI onset, exclusion of competing causes, and outcome available for review. Retrospective study patients were either interviewed in person or over the phone to review the dose and duration of suspect medication use and facilitate collection of a DNA sample after obtaining written informed consent.
DILIGEN study
Only Caucasian subjects with DILI attributed to minocycline enrolled in the DILIGEN study with an available DNA sample were included. All subjects had (a) clinically apparent jaundice or bilirubin > 40 umol/L, or (b) a serum ALT > 5 × ULN or (c) Alk P > 2 × ULN plus any raised bilirubin above ULN (7). All patients had a Roussel Uclaf Causality Assessment Method (RUCAM) causality score of 3 or greater.
Liver histopathology
Available liver biopsies were reviewed by a single expert liver histopathologist (DEK). All samples were scored for multiple histological features as well as an overall pattern of liver injury (11).
Causality assessment
The causal relationship between the liver injury episode and the minocycline use were evaluated in a standardized fashion by the DILIN causality committee (12). A DILIN expert opinion causality score varying from 1 (Definite > 95% likelihood), 2 (Highly Likely 75%-95% likelihood), 3 (Probable 50%-74% likelihood), 4 (Possible 25%-49% likelihood) to 5 (unlikely < 25% likelihood) was assigned by consensus agreement of committee members for all of the retrospective and prospective DILIN cases. In addition, a RUCAM score was calculated for each case and implicated agent (13). In subjects with 2 or more implicated drugs, an overall causality score was assigned to the case and then an individual causality score for each drug was given.
Controls
Since DILI has a very low prevalence and minocycline is widely prescribed in healthy individuals with an estimated 4.2 million prescriptions of tetracylines per year in the US, unselected population samples were used as study controls (14). We selected 10,588 Caucasian controls from different available sources; the Welcome Trust Case Control Consortium (WTCCC) (http://www.wtccc.org.uk), the population reference sample (POPRES)and PGX4000118 and Spanish Bladder cancer cohort from dbGAP
A
uthor Man
uscr
ipt
A
uthor Man
uscr
ipt
A
uthor Man
uscr
ipt
A
uthor Man
uscr
(phs000346.v1). Since all cases of minocycline DILI were determined to be of primarily Northern European ancestry, the set of ancestry-matched controls totaled 6,835 individuals. Since prior medication exposure history was not available for the controls, we presume that none of these patients previously received minocycline.
Genome-wide SNP and HLA analysis
Genome-wide genotyping of DILI cases was performed by the Broad Institute in Boston by Illumina Infinium HumanCoreExome BeadChip (n=19) or at the Duke Center for Human Genome Variation on the Illumina 1Mduo array (n=6). A total of 505,740 markers shared across the different genotyping platforms used for DILI cases and controls passed quality control (QC) and no samples were excluded for low quality profile.
For each cohort, single nucleotide polymorphisms (SNPs) with poor quality data were pruned before the imputation to avoid false positives. The imputation was performed by batches dividing the cohorts by the genotyping platform used. For each batch, we first phased the data by SHAPEIT (version v2.r727)(15), to increase the accuracy of the imputation. Then, imputation was carried out using IMPUTE2 (version 3) with the 1000 Genomes Project (release v321) dataset as the reference panel (16). We used an ethnically mixed panel to improve the quality of the imputation for rare variants. We retained imputed genotypes with: (a) posterior probability > 0.9 in each genotyping batch, (b) no significant difference in missingness between cases and controls (χ2 test, p-value > 0.0001), (c) no significant deviation from Hardy-Weinberg equilibrium (p-value > 0.0001), (d) missing data not greater than 5% in each single genotyping batch, (e) info score greater than 0.8 in each genotyping batch. Since the imputation quality is higher for common variants, we selected SNPs with MAF in the 1000 Genomes Project greater or equal of 0.01. The imputed cohorts were then merged and genotyped SNPs were used to replace imputed SNP genotypes if previously eliminated during the build of the batch groups. For each imputed SNP, possible batch effects were detected by testing the association (by logistic regression) between controls within the same population typed by different platforms. SNPs with association p-values less than 0.005 in this control comparison were excluded from the analysis. For each cohort, HLA alleles were also inferred using HIBAG (17) using the reference predictor panels specific for each genotyped chip. The top associated HLA type was further validated by sequence-based HLA typing at Histogenetics (Ossining, NY, USA). A proxy for HLA-B*35:02, rs148631562, was typed with a TaqMan® SNP genotyping assay (ThermoFisher Scientific, Waltham, MA) in accordance with the manufacturer's recommendations in 7 additional European ethnicity cases.
Statistical analysis
The effect of population structure was assessed through principal components analysis (PCA) using the smartPCA program from the EIGENSTRAT package (version 3.0) (18). The statistical association of each marker (HLA allele or SNP) with risk for minocycline DILI was determined by logistic regression with the first ten significant principal
components as covariates under an additive genetic model. The lack of association between the HLA-B*35:02 allele and EIGENSTRAT axes can be seen in Figure S1. Association and
A
uthor Man
uscr
ipt
A
uthor Man
uscr
ipt
A
uthor Man
uscr
ipt
A
uthor Man
uscr
heterogeneity tests were carried out using PLINK (19). We set the genome-wide significance p-value threshold to 1.0 × 10-8 to correct for multiple testing. Given the high prior
probability of HLA association with risk for DILI, an HLA-wide significance threshold was determined by Bonferroni correction for the 217 HLA alleles tested for association (p=2.3 × 10-4). No test statistic inflation was observed for the results. To test for independent effects within regions having multiple associated variants, we included one or more variants as covariates and tested the residual effect of these variants after adjusting for the top
associated allele. Differences in clinical characteristics among sample groups were tested by Fisher's Exact test or one-way ANOVA. All detailed analyses and Manhattan plots were performed with R (Version 3.0.2). Regional plots were drawn by LocusZoom (20).
HLA modelling and molecular docking
An atomic model of B*35:02 was generated by SWISS-MODEL (21) using the HLA-B*35:02 sequence found in IMGT/HLA (22). The SMILES string of minocycline was obtained from PubChem and translated into 3 dimensional coordinates using the NCI/CADD translator (http://cactus.nci.nih.gov/translate/). AutoDock Tools was used to prepare files for molecular docking using AutoDock Vina (23). The top 9 scoring orientations were studied with the highest scoring pose shown in Figure 3.
Results
Patient population
A total of 25 Caucasian patients with DILI attributed to minocycline constitute our study population. Eighteen of the patients were enrolled in the DILIN prospective study, 4 were recruited in the DILIN retrospective study and 3 subjects were enrolled from DILIGEN. As shown in Table 1, the majority of subjects were female (80%) with a median age of 19 years (range: 16-61). Indications for minocycline use were dermatologic in 100%. At presentation, the majority of subjects had an acute hepatocellular pattern of liver injury with a median serum ALT of 1077 U/L, median Alk P of 176 U/L, and 52% of the subjects were jaundiced. The median duration of minocycline use was 55 weeks (range: < 1 to >122) with 52% having taken the drug for > 12 months. Hypersensitivity features at presentation included 4% with eosinophilia, 32% with fever, and 20% with a rash. The majority of subjects had a + ANA (90%) determined at their local hospital lab, 32% had detectable smooth muscle antibody, and 58% had hypergammaglobulinemia.
A liver biopsy was obtained during the process of clinical evaluation and available for central review in 8 subjects (Table 2). The time from DILI onset to liver biopsy varied from 2 to 220 days with a median time of 73 days. The pattern of injury was acute (lobular predominant) hepatitis in 3 patients and chronic (portal predominant) hepatitis in 5 patients. In 2 of the cases of acute hepatitis and one of the cases of chronic hepatitis, there was mild to moderate zone 3 bile accumulation (cholestatic hepatitis). Half of the cases had marked interface hepatitis and in 2 cases, this was associated with bridging necrosis. Mild confluent necrosis was seen in 3 cases. Portal plasma cell infiltrates were prominent in 3 cases, while eosinophils were prominent in 5 cases. The 3 cases of cholestatic hepatitis all had prominent portal neutrophil infiltrates. On the basis of pattern and plasma cell infiltration only 3 cases
A
uthor Man
uscr
ipt
A
uthor Man
uscr
ipt
A
uthor Man
uscr
ipt
A
uthor Man
uscr
showed classic features that would suggest autoimmune hepatitis. Fibrosis was present as periportal expansion in most cases, but two cases had evidence of early bridging fibrosis.
During follow-up, none of the patients died or underwent liver transplantation. However, 5 (28%) of the 18 DILIN prospective patients met pre-determined criteria for chronic DILI at 6 months after injury onset. At 24 months of follow-up, all 5 patients had normalized their labs. Local physicians treated 12 subjects (50%) with corticosteroids. The median duration of steroid use was 160 days (range: 4 to 435 days) and 27% of the DILIN patients remained on steroids for at least 6 months after DILI onset.
Genetic association studies
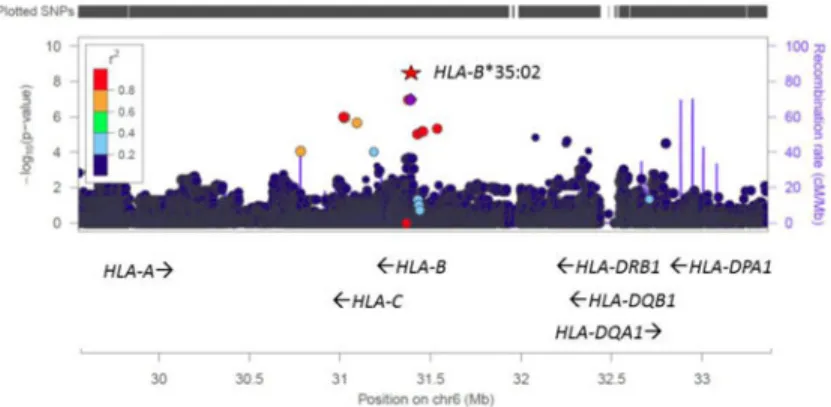
A genome-wide association study was undertaken on the 25 minocycline DILI cases, including association testing of HLA alleles imputed from SNP genotype data in the major histocompatibility complex (MHC) region on chromosome 6. The results of the genome-wide scan did not reveal any individual SNP or genomic region showing significant association with risk for minocycline-DILI after multiple test correction. However, when assessing the HLA types imputed from SNP genotype data in the MHC region, we observed a significant enrichment of the HLA-B*35:02 allele in the cases versus population controls (16% vs 0.6%, Odds ratio 29.6, 95% CI: 7.8-89.8, p = 2.5 × 10-8) (Table 3). The association with the imputed HLA-B*35:02 allele was then confirmed with sequence-based HLA typing (Figures 1 and 2).
HLA docking studies
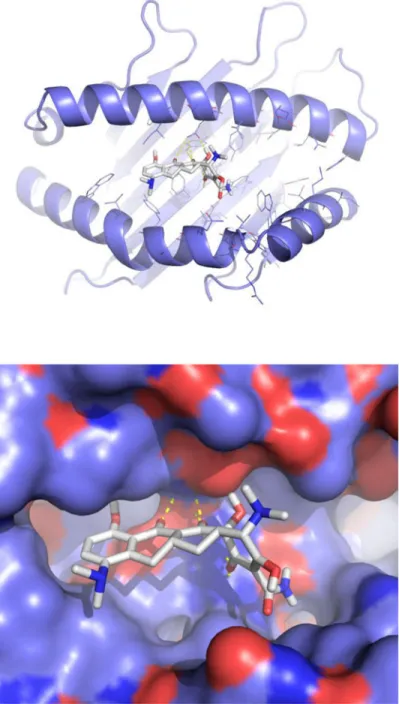
Although the mechanistic basis for HLA associations with DILI is not known, it is presumed to involve: 1) direct binding of either the drug or a drug metabolite to HLA molecules or a metabolite-peptide adduct binding to HLA molecules, 2) effects on selection (expansion or deletion) of HLA restricted drug-specific T cells, and/or 3) indirect effects due to linked causative genes. To determine if minocycline has the potential to bind HLA-B*35:02, we used molecular docking to predict putative interactions. Although the structure of HLA-B*35:02 has not been solved, it is sufficiently similar (98.9 %) to a solved crystal structure (HLA-B*35:01, PDB code 1XH3) allowing the generation of a high confidence model. The crystal structure of abacavir bound to HLA-B*57:01 provided a rational basis for prediction of sites within the antigen binding cleft that permit drug/HLA interactions (24, 25) . We used a high confidence model of HLA-B*35:02 and knowledge of drug/HLA contact sites to predict the likelihood of direct minocycline interactions with HLA-B*35:02. We used AutoDock Vina to dock minocycline into the antigen binding cleft of HLA-B*35:02 yielding a predicted interaction (estimated ΔG=-7.9 kcal per mole, compared to control docking of abacavir into HLA-B*57:01, ΔG=-7.2 kcal per mole (Figure 3). These data suggest that minocycline has the potential to bind the HLA-B*35:02 antigen binding cleft.
Discussion
Our study demonstrates a higher frequency of HLA-B*35:02 amongst the 25 patients with minocycline DILI compared to a large group of population controls. The GWAS results were confirmed using sequence-based HLA typing. Although further validation in an independent
A
uthor Man
uscr
ipt
A
uthor Man
uscr
ipt
A
uthor Man
uscr
ipt
A
uthor Man
uscr
and larger group of patients with minocycline DILI are needed, the results are in line with previous genetic studies of DILI showing (often drug-specific) risk factors in both HLA Class I and HLA Class II genes (5,6). HLA-B*35:02 has a low frequency in the general population, with an allele frequency of only 0.3% in Caucasians and less than 0.1% in African Americans.
A review of the literature demonstrates that HLA-* 35:02 is moderately associated with the rate of HIV progression to AIDS in Caucasian patients (26). It remains possible that HLA*B-35:02 could be a marker for the underlying condition of facial acne for which minocycline was prescribed. However, prior GWAS in patients with acne has failed to demonstrate this association (27-29). Furthermore, our molecular docking data suggest that minocycline is capable of binding within the HLA-B*35:02 antigen binding cleft, and may potentially alter the profile of peptides that normally bind to HLA-B*35:02, as has been shown with abacavir binding to HLA-B*57:01 (30). Crystal structures demonstrate that abacavir forms contact with specific peptides and HLA-B*57:01. Several contacts between the drug, peptide and HLA are mediated by ordered water molecules. Molecular docking of minocycline to HLA-B*35:02 is limited by lack of knowledge regarding peptides that bind in the presence of the drug. Moreover, it is not possible to accurately predict the locations of ordered water molecules in a model of HLA-B*35:02. Future studies characterizing peptides bound to HLA-B*35:02 are expected to elucidate functional drug interaction mechanisms for this (and other) HLA-DILI associations.
No significant difference in clinical presentation or outcome based upon HLA-B* 35:02 genotype was observed except for lower total bilirubin levels and R-values (Table 1) (see supplemental Table 1 for additional information). This may be due to the limited number of cases enrolled (25), with only 4 HLA-B*35:02 carriers among this set, or a true lack of difference in presentation or outcomes related to HLA-B*35:02 carrier status. A larger number of minocycline DILI cases used for genetic validation could also help determine if the HLA-B*35:02 carriers have any unique clinical or laboratory features compared to non-carriers with minocycline DILI. Nonetheless, our data are consistent with other GWAS studies in DILI subjects demonstrating an association of HLA region with DILI
susceptibility (31-33). Many of these are in the HLA Class I genes (specifically, HLA-A or HLA-B), though the HLA Class II genes have also been implicated. These studies suggest a role for adaptive immunity in DILI pathogenesis.
Many of our patients had autoimmune like features at presentation. Furthermore, available liver biopsies demonstrated some features of AIH in some patients (Table 2). Up to 50% of patients were treated with steroids and most did well. Steroids were successfully tapered off in most patients. Although there were no liver transplants or deaths, 24% evolved into chronic DILI which is higher than our prior report (9). This may in part be due to prolonged use of the drug in these patients. Furthermore, most of these patients did not get routine labs drawn prior to use. Lastly, a substantial delay between first evidence of DILI (abnormal labs) and drug cessation was observed with 11 subjects discontinuing the drug after a median of 38 days after DILI onset. Prior studies have suggested that continued suspect drug use is associated with more severe DILI and chronicity (34). However, the median duration of minocycline use in the 5 DILIN subjects who developed chronic DILI was not significantly
A
uthor Man
uscr
ipt
A
uthor Man
uscr
ipt
A
uthor Man
uscr
ipt
A
uthor Man
uscr
longer than the duration of use in the 12 without chronic DILI (252 vs 400 days, p=0.6). And the duration of minocycline use after DILI onset was not longer in those with chronic vs self-limited DILI (1 day for both groups, p=0.8). Fortunately at month 24 of follow-up, none of the 5 chronic DILI patients had evidence of cirrhosis, portal hypertension or liver failure and in fact all had normalized their liver biochemistries.
Recent studies have suggested that certain HLA alleles (i.e. DRB1*0301, HLA-DRB1*0401) may be associated with susceptibility to autoimmune hepatitis in Caucasians (35). When we looked for these specific alleles, there was no over-representation in our minocycline cases compared to population controls (Data not shown). However, the lack of an association may be due to lack of power owing to the small number of minocycline DILI cases.
Given the low incidence of DILI amongst patients receiving minocycline, HLA typing is unlikely to help prevent minocycline DILI. However, HLA-B*35:02 may be useful as a diagnostic aid in the setting of suspected minocycline DILI especially in distinguishing it from sporadic autoimmune hepatitis but confirmatory studies are needed. In addition due to the small number of case patients that carry the HLA-B*35:02 allele (n=4), validation of these findings in a larger, independent cohort of patients with minocycline DILI is needed.
The mechanism linking minocycline to HLA-B*35:02 is not clear. One possibility is that the drug binds directly to the antigen binding cleft similar to abacavir binding to HLA-B*57:01, and alters the repertoire of bound 9mer peptide ligands- presumably presenting novel cell surface antigens. However, minocycline (molecular weight 493.9 daltons) is larger than abacavir (molecular weight 286.3 daltons) and may hinder conventional 9mer peptide ligand interactions. That is, minocycline may be presented in the absence of conventional peptides (e.g., 9mers). An alternative model is that minocycline metabolites bind HLA, forming complexes recognized by CD8+ cytotoxic T-lymphocytes. Another potential mechanism is that stressed/damaged hepatocytes exhibit immunogenic peptide/HLA complexes that act as neoantigens. It also remains possible that minocycline or a metabolite binds covalently to cellular proteins resulting in the presentation of a drug-peptide complex to T-cells via HLA-B* 35:02, as appears to occur with flucloxacillin in HLA-B * 57:01 carriers (36). Evidence for the formation of a reactive intermediate(s) from minocycline but not other tetracyclines has been reported previously (37). Our group and others are planning additional in vitro experiments in patients with and without the HLA-B * 35:02 allele to follow-up on these preliminary studies. Elucidating the mechanism(s) underlying the association between HLA-B*35:02 and risk for minocycline hepatotoxicity will lead to a greater understanding of the pathophysiology of liver injury from minocycline, and perhaps other hepatotoxic drugs.
Supplementary Material
Refer to Web version on PubMed Central for supplementary material.
A
uthor Man
uscr
ipt
A
uthor Man
uscr
ipt
A
uthor Man
uscr
ipt
A
uthor Man
uscr
Acknowledgments
DILIN Clinical Sites: Indiana University-Purdue: Naga Chalasani, MD, PI; Marwan S. Ghabril, MD, Sub-I; Suthat Liangpunsakul, MD, Sub-I; Raj Vuppalanchi, MD, Sub-I; [Audrey Corne, RN, EdD, Study Coord; Sherrie Cummings, RN, BSN, Study Coord; Wendy Morlan, RN, Study Coord];
University of Michigan-Ann Arbor: Robert J. Fontana, MD, PI; Hari Conjeevaram, MD, Sub-I; Frank DiPaola, MD, Sub-I; [Cassandra Coffman, Study Coord; Sophana Mao, Study Coord];
University of North Carolina-Chapel Hill: Paul Watkins, MD, PI; Jama Darling, MD, I; Michael Fried, Sub-I; Paul H. Hayashi, MD, Sub-Sub-I; Steven Lichtman, MD, Sub-Sub-I; Steven Zacks, MD, MPH, Sub-Sub-I; [Tracy Russell, CCRP, Study Coord];
Satellite Sites: Asheville: William Harlan, MD, PI; [Tracy Russell, CCRP, Study Coord];
Wake Forest Baptist Medical Center: Herbert Bonkovsky, MD, PI; Pradeep Yarra, MD, Sub-I; [Denise Faust, Study Coord];
University of Southern California: Andrew Stolz, MD, PI; Neil Kaplowitz, MD, I; John Donovan, MD, Sub-I; [Susan Milstein, RN, BSN, Study Coord];
Satellite Sites: University of California-Los Angeles (Pfleger Liver Institute): Francisco A. Durazo, MD, PI; [Yolanda Melgoza, Study Coord; Val Peacock, RN, BSN, Co-Coord]
Albert Einstein Medical Center: Victor J. Navarro, MD, PI; Simona Rossi, MD, Sub-I; [Maricruz Vega, MPH, Study Coord; Manisha Verma, MD, MPH, Study Coord]
Icahn School of Medicine at Mount Sinai: Joseph Odin, MD, PhD, PI; Jawad Ahmad, MD, PI; Nancy Bach, I; Meena Bansal, MD, I; Charissa Chang, MD, I; Douglas Dieterich, MD, I; Priya Grewal, MD, Sub-I; Lawrence Liu, MD, Sub-Sub-I; Thomas Schiano, MD, Sub-Sub-I; [Monica Taveras, Study Coord]
National Institutes of Health Clinical Center: Christopher Koh, MD, PI; [Beverly Niles, Study Coord]
DILIN Data Coordinating Center at Duke Clinical Research Institute: Huiman X. Barnhart, PhD, PI; Katherine Galan, RN, Project Lead; Theresa O'Reilly, Lead CRA; Elizabeth Mocka, CRA; Olivia Pearce, CTA; Michelle McClanahan-Crowder, Data Management; Coleen Crespo-Elliott, Data Management; Hoss Rostami, Data Management; Qinghong Yang, Programmer-Statistics; Jiezhun (Sherry) Gu, PhD, Statistician; Tuan Chau, Lead Safety Associate; Liz Cirulli-Rogers, PhD, Pharmacogenetics statistician
National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK): José Serrano, MD, Project Scientist; Rebecca J. Torrance, RN, MS, Clinical Trials Specialist; Rebekah Van Raaphorst, MPH, LT, USPHS, Health Research Administrator; Francisco O. Calvo, PhD, COC Contact; James E. (Jay) Everhart, MD, MPH, Scientific Advisor; Jay Hoofnagle, MD, Scientific Advisor; Averell H. Sherker, MD, FRCP(C), Program Official.
Financial support: The DILIN Network is structured as a U01 cooperative agreement with funds provided by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) under grants: 2U01-DK065176-06 (Duke) , 2U01-DK065201-06 (UNC), 2U01-DK065184-06 (Michigan), 2U01-DK065211-06 (Indiana), 5U01DK065193-04 (UConn) , 5U01-DK065238-08 (UCSF/CPMC), DK083023-01 (UTSW), 1U01-DK083027-01 (TJH/UPenn), 1U01-DK082992-01 (Mayo), 1U01-DK083020-01 (USC). Additional funding is provided by CTSA grants: UL1 RR025761 (Indiana), UL1TR000083 (UNC), UL1 RR024134 (UPenn), UL1 RR024986 (Michigan), UL1 RR024982 (UTSW), UL1 RR024150 (Mayo) and in part by the Intramural Research Program of the NIH, National Cancer Institute. This study was also supported in part by DK089464 (T.J.U). In addition, the International Serious Adverse Events Consortium (iSAEC) provided funding for collection of biological samples and data from European cases. Finally, the National Institute for Health Research in the United Kingdome provided support via the Nottingham Digestive Diseases Biomedical Research unit (GPA).
Dr. Fontana has received research support from BMS and Gilead and served as a consultant to Regulus, Abbvie, and Alnylam.
Naga Chalasani, MD, FAASLD has ongoing consulting agreements related to NAFLD and NASH from Lilly, Tobira, Allergan, Celgene, NuSirt, DS Biopharma, Shire, and Madrigal. He previously had consulting agreements with Mitsubishi-Tanabe and Boeringher-Ingelheim. Over the last 3 years, he has received compensation for providing consultation related to drug hepatotoxicity from Cempra, Abbvie, Merck, and Salix. Dr. Chalasani has served as the site Principal Investigator for clinical trials funded by Intercept, Gilead and Galectin where his institution was the recipient of the funding.
A
uthor Man
uscr
ipt
A
uthor Man
uscr
ipt
A
uthor Man
uscr
ipt
A
uthor Man
uscr
References
1. Ostapowicz G, Fontana RJ, Schiodt FV, Larson A, Davern TJ, Han SH, et al. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann Intern Med. 2002; 137:947–954. [PubMed: 12484709]
2. Bjornsson ES, Bergmann OM, Bjornsson HK, Kvaran RB, Olafsson S. Incidence, presentation, and outcomes in patients with drug-induced liver injury in the general population of Iceland.
Gastroenterology. 2013; 144:1419–1425. 1425 e1411–1413. quiz e1419-1420. [PubMed: 23419359] 3. Chalasani N, Fontana RJ, Bonkovsky HL, Watkins PB, Davern T, Serrano J, et al. Causes, clinical
features, and outcomes from a prospective study of drug-induced liver injury in the United States. Gastroenterology. 2008; 135:1924–1934. 1934 e1921–1924. [PubMed: 18955056]
4. Bjornsson E, Talwalkar J, Treeprasertsuk S, Kamath PS, Takahashi N, Sanderson S, et al. Drug-induced autoimmune hepatitis: clinical characteristics and prognosis. Hepatology. 2010; 51:2040– 2048. [PubMed: 20512992]
5. Lucena MI, Molokhia M, Shen Y, Urban TJ, Aithal GP, Andrade RJ, et al. Susceptibility to
Amoxicillin-Clavulanate-Induced Liver Injury is Influenced by Multiple HLA Class I and II Alleles. Gastroenterology. 2011
6. Singer JB, Lewitzky S, Leroy E, Yang F, Zhao X, Klickstein L, et al. A genome-wide study identifies HLA alleles associated with lumiracoxib-related liver injury. Nat Genet. 2010; 42:711– 714. [PubMed: 20639878]
7. Spraggs CF, Budde LR, Briley LP, Bing N, Cox CJ, King KS, Whittaker JC, et al. HLA-DQA1*02:01 Is a Major Risk Factor for Lapatinib-Induced Hepatotoxicity in Women With Advanced Breast Cancer. J Clin Oncol. 2011; 29:667–673. [PubMed: 21245432]
8. Daly AK, Donaldson PT, Bhatnagar P, Shen Y, Pe'er I, Floratos A, Daly MJ, et al. HLA-B*5701 genotype is a major determinant of drug-induced liver injury due to flucloxacillin. Nat Genet. 2009; 41:816–819. [PubMed: 19483685]
9. Fontana RJ, Watkins PB, Bonkovsky HL, Chalasani N, Davern T, Serrano J, Rochon J. Drug-Induced Liver Injury Network (DILIN) prospective study: rationale, design and conduct. Drug Saf. 2009; 32:55–68. [PubMed: 19132805]
10. DeBoer YS, Kosinski AS, Urban TJ, Zhao Z, Long N, Chalasani N, et al. Features of Autoimmune hepatitis in Patients with Drug Induced Liver Injury. Clin Gastro Hep. 2017; 15:103–112.
11. Kleiner DE, Chalasani NP, Lee WM, Fontana RJ, Bonkovsky HL, Watkins PB, Hayashi PH, et al. Hepatic histological findings in suspected drug-induced liver injury: systematic evaluation and clinical associations. Hepatology. 2014; 59:661–670. [PubMed: 24037963]
12. Rockey DC, Seeff LB, Rochon J, Freston J, Chalasani N, Bonacini M, Fontana RJ, et al. Causality assessment in drug-induced liver injury using a structured expert opinion process: comparison to the Roussel-Uclaf causality assessment method. Hepatology. 2010; 51:2117–2126. [PubMed: 20512999]
13. Benichou C, Danan G, Flahault A. Causality assessment of adverse reactions to drugs--II. An original model for validation of drug causality assessment methods: case reports with positive rechallenge. J Clin Epidemiol. 1993; 46:1331–1336. [PubMed: 8229111]
14. Hicks LA, Bartoces MG, Roberts RM, Suda KJ, Hunkler RJ, Taylor TH, et al. US outpatient Antibiotic Prescribing variation according to geography, Patient population, and Provider Speciality in 2011. Clin Inf Dis. 2015; 60:1308–1316.
15. Delaneau O, Marchini J, Zagury JF. A linear complexity phasing method for thousands of genomes. Nat Methods. 2012; 9:179–181.
16. Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009; 5:e1000529. [PubMed: 19543373]
17. Zheng X, Shen J, Cox C, Wakefield JC, Ehm MG, Nelson MR, Weir BS. HIBAG--HLA genotype imputation with attribute bagging. Pharmacogenomics J. 2014; 14:192–200. [PubMed: 23712092] 18. Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components
analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006; 38:904– 909. [PubMed: 16862161]
A
uthor Man
uscr
ipt
A
uthor Man
uscr
ipt
A
uthor Man
uscr
ipt
A
uthor Man
uscr
19. Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007; 81:559–575. [PubMed: 17701901]
20. Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP, Boehnke M, et al.
LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics. 2010; 26:2336–2337. [PubMed: 20634204]
21. Schwede T, Kopp J, Guex N, Peitsch MC. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 2003; 31:3381–3385. [PubMed: 12824332]
22. Robinson J, Halliwell JA, Hayhurst JD, Flicek P, Parham P, Marsh SG. The IPD and IMGT/HLA database: allele variant databases. Nucleic Acids Res. 2015; 43:D423–431. [PubMed: 25414341] 23. Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. AutoDock4
and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem. 2009; 30:2785–2791. [PubMed: 19399780]
24. Ostrov DA, Grant BJ, Pompeu YA, Sidney J, Hamdahl M, Southwood S, et al. PNAS. 2012; 109:9959–9964. [PubMed: 22645359]
25. Illing PT, Vivian JP, Dudek NL, Kostenko L, Chen Z, Bharadwai M, et al. Immune self-reactivity triggered by drug-modified JLA-peptide repetoire. Nature. 2012; 486:554–558. [PubMed: 22722860]
26. Gao X, Nelson GW, Karacki P, Martin MP, Phair J, Kaslow R, Goedert JJ, et al. Effect of a single amino acid change in MHC class I molecules on the rate of progression to AIDS. N Engl J Med. 2001; 344:1668–1675. [PubMed: 11386265]
27. He L, Wu WJ, Yang JK, Cheng H, Zuo XB, Lai W, Gao TW, et al. Two new susceptibility loci 1q24.2 and 11p11.2 confer risk to severe acne. Nat Commun. 2014; 5:2870. [PubMed: 24399259] 28. Navarini AA, Simpson MA, Weale M, Knight J, Carlavan I, Reiniche P, Burden DA, et al.
Genome-wide association study identifies three novel susceptibility loci for severe Acne vulgaris. Nat Commun. 2014; 5:4020. [PubMed: 24927181]
29. Zhang M, Qureshi AA, Hunter DJ, Han J. A genome-wide association study of severe teenage acne in European Americans. Hum Genet. 2014; 133:259–264. [PubMed: 24114350]
30. Ostrov DA, Grant BJ, Pompeu YA, Sidney J, Harndahl M, Southwood S, Oseroff C, et al. Drug hypersensitivity caused by alteration of the MHC-presented self-peptide repertoire. Proc Natl Acad Sci U S A. 2012; 109:9959–9964. [PubMed: 22645359]
31. Daly AK. Drug-induced liver injury: past, present and future. Pharmacogenomics. 2010; 11:607– 611. [PubMed: 20415545]
32. Urban TJ, Goldstein DB, Watkins PB. Genetic basis of susceptibility to drug-induced liver injury: what have we learned and where do we go from here? Pharmacogenomics. 2012; 13:735–738. [PubMed: 22594502]
33. Urban TJ, Daly AK, Aithal GP. Genetic basis of drug-induced liver injury: present and future. Semin Liver Dis. 2014; 34:123–133. [PubMed: 24879978]
34. Aithal GP, Watkins PB, Andrade RJ, Larrey D, Molokhia M, Takikawa H, Hunt CM, et al. Case definition and phenotype standardization in drug-induced liver injury. Clin Pharmacol Ther. 2011; 89:806–815. [PubMed: 21544079]
35. de Boer YS, van Gerven NM, Zwiers A, Verwer BJ, van Hoek B, van Erpecum KJ, Beuers U, et al. Genome-wide association study identifies variants associated with autoimmune hepatitis type 1. Gastroenterology. 2014; 147:443–452. [PubMed: 24768677]
36. Monshi MM, Faulkner L, Gibson A, Jenkins RE, Farrell J, Earnshaw CJ, et al. Human leukocyte antigen (HLA)-B*57:01-restricted activation of drug-specific T cells provides the immunological basis for flucloxacillin-induced liver injury. Hepatology. 2012; 57:727–739.
37. Mannargudi B, McNally D, Reynolds W, Uetrecht J. Bioactivation of minocycline to reactive intermediates by myeloperoxidase, horseradish peroxidase, and hepatic microsomes: implications for minocycline-induced lupus and hepatitis. Drug Metab Dispo. 2009; 37(9):1806–18.
A
uthor Man
uscr
ipt
A
uthor Man
uscr
ipt
A
uthor Man
uscr
ipt
A
uthor Man
uscr
Abbreviations
ALK P Alkaline phosphatase
ALT Alanine aminotransferase
AST Aspartate aminotransferase
DILI Drug induced liver injury
DILIGEN Drug induced liver injury genetics group
DILIN Drug induced liver injury network
GWAS Genome wide association study
HLA Human leukocyte antigen
INR International normalized ratio
MAF Minor allele frequency
MHC Major histocompatibility complex
RUCAM Rousell Uclaf Causality assessment method
SNP Single nucleotide polymorphism
ULN Upper limit of normal
A
uthor Man
uscr
ipt
A
uthor Man
uscr
ipt
A
uthor Man
uscr
ipt
A
uthor Man
uscr
Lay summary
Development of liver injury following prolonged use of minocycline for acne is a rare but potentially severe form of drug induced liver injury. Our study demonstrates that
individuals who are HLA-B *35:02 carriers are at increased risk of developing minocycline related liver injury. These results may help doctors more rapidly and confidently diagnose affected patients and possibly reduce the risk of liver injury in individuals receiving minocycline going forward.
A
uthor Man
uscr
ipt
A
uthor Man
uscr
ipt
A
uthor Man
uscr
ipt
A
uthor Man
uscr
Figure 1. Manhattan plot showing genome-wide single variant association test results
The -log10(P value) from a logistic regression of each SNP on phenotype is plotted according to physical location of the SNPs on each of the 22 autosomes, with SNPs on different chromosomes colored with alternating colors. SNPs that exceeded a p-value of 10-5 threshold are marked in green color.
A
uthor Man
uscr
ipt
A
uthor Man
uscr
ipt
A
uthor Man
uscr
ipt
A
uthor Man
uscr
Figure 2. LocusZoom plot of association test results in the MHC region
The -log10(P value) from a logistic regression of each SNP as well as HLA-B*35:02 on phenotype is plotted according to physical locations in the MHC region. Linkage disequilibrium between each of the SNPs and the lead GWAS SNP (rs146765245) is indicated by color. Associations with the GWAS SNPs were not genome-wide significant, but imputation of HLA types revealed a genome-wide significant association with HLA-B*35:02. Local recombination rate in this region is also shown with the second Y-axis on the right. Several HLA genes with their direction of transcription are marked by arrows on the bottom and the locations of SNPs are marked by vertical bars on the top. This plot is generated using the web tool LocusZoom at http://locuszoom.sph.umich.edu/locuszoom/.
A
uthor Man
uscr
ipt
A
uthor Man
uscr
ipt
A
uthor Man
uscr
ipt
A
uthor Man
uscr
Figure 3. Molecular docking of minocycline in the HLA-B*35:02 antigen binding cleft
(a) A ribbon diagram of HLA-B*35:02 showing minocycline with white sticks for carbon, blue for nitrogen, red for oxygen, and white for hydrogen. Yellow dashes represent polar interactions between minocycline and HLA-B*35:02. (b). Minocycline is shown in the top scoring binding orientation predicted by molecular docking using AutoDock Vina. The molecular surface of HLA-B*35:02 is showing as violet for carbon, blue for nitrogen, red for oxygen, and white for hydrogen.
A
uthor Man
uscr
ipt
A
uthor Man
uscr
ipt
A
uthor Man
uscr
ipt
A
uthor Man
uscr
A
uthor Man
uscr
ipt
A
uthor Man
uscr
ipt
A
uthor Man
uscr
ipt
A
uthor Man
uscr
ipt
T ab le 1 Pr esenting features and outcomes of 25 patients with minocycline hepatotoxicity
Characteristic
n
T
otal Sample
HLA-B*35:02 (-) N=21
HLA B-35:02 (+) N=4
p-v
alue
Demographics Age, median (interquartile range)
25 19 (17.9-41.0) 19.5 (17.3-41.0) 19.6 (19.5-30.2) 0.96 % Female 25 80% 81% 75% 1
BMI (mean +/- SD)
21
22.9 +/- 3.9
22.7 +/- 4.2
23.8 +/- 2.6
0.56
Clinical F
eatur
es
Latenc
y (days) (mean +/- SD)
24
346 +/- 268
387 +/- 249
416 +/- 376
0.69
R-v
alue at onset (mean +/- SD)
25
15.6 +/- 11.5
17.6 +/- 12.8
8.4 +/- 4.8
0.030
AL
T (U/L) at onset, median (25
th, 75 th)
22
1111(278, 1996)
1474 (278, 2009)
545 (343, 1169)
0.50
Bilirubin (mg/dL) at onset, median (25
th, 75 th)
22
4.5 (0.5, 7.6)
4.9 (0.7, 7.7)
0.4 (0.3, 3.6)
0.05 Immunoaller gic features: Fe v er 25 8/25 (32%) 7/21 (33%) 1/4 (25%) 1 Rash 25 5/25 (20%) 5/21 (24%) 0/4 (0%) 0.55 Eosinophilia 23 1/23 (4.3%) 1/19 (5.3%) 0/4 (0%) 1 +AN A 21 19/21 (90%) 4/4 (100%) 15/17 (88%) 1 +SmAb 21 7/21 (33%) 6/17 (35%) 1/4 (25%) 1 Recei v ed Corticosteroids 22 11/22 (50%) 9/18 (50%) 2/4 (50%) 1 Chronic DILI 21 5/17 (24%) 3/17 (18%) 2/4 (50%) 0.23 DILIN Se v
erity Score, median (25
th, 75 th)
22
2.0 (1.0, 2.0)
2.0 (1.0, 2.0)
1.0 (1.0, 2.0)
A
uthor Man
uscr
ipt
A
uthor Man
uscr
ipt
A
uthor Man
uscr
ipt
A
uthor Man
uscr
ipt
Table 2
Liver histopathology in 8 DILIN subjects with minocycline hepatotoxicity
Age (years) Gender
Duration of Minocycline use (d)
Days from DILI
onset to biopsy Findings
17 Male 570 144 Marked chronic hepatitis, eosinophils
18 Female 701 22
Marked chronic hepatitis, plasma cells and eosinophils, bridging fibrosis
16 Female 197 101 Moderate chronic hepatitis
55 Female 588 8 Moderate cholestatic chronic hepatitis, plasma cells, eosinophils
17 Female 387 220 Mild chronic hepatitis, plasma cells
31 Female 43 10 Marked acute cholestatic hepatitis, plasma cells, eosinophils
19 Female 74 5 Marked acute hepatitis, plasma cells, early bridging fibrosis
A
uthor Man
uscr
ipt
A
uthor Man
uscr
ipt
A
uthor Man
uscr
ipt
A
uthor Man
uscr
ipt
T
ab
le 3
Association test r
esults f
or top HLA allele and top SNP in MHC r
egion
MAF (cases)
MAF (contr
ols)
Carrier Fr
equency (cases, N = 25)
Carrier Fr
equency (contr
ols, N= 6,835)
Odds Ratio (95% CI)
p-v
alue
HLA-B*35:02
8%
0.3%
16.0%
0.6%
29.6 (7.8, 89.8)
2.57 × 10
-8
rs146765245
10%
0.2%
16.0%
0.4%
29.9 (8.1, 97.6)
1.01 × 10
-7
MAF= minor allele frequenc