Using Maximum Entropy to Extract Biomedical Named Entities without Dictionaries
Full text
Figure

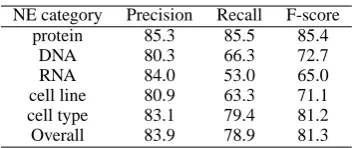
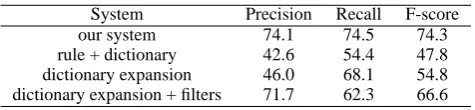
Related documents
The main findings of this study have generated some important obstacles which have been faced by jute growers of Chandpur Districts such as lack of capital and inputs,
Radiographic data before the sixth lengthening surgery were measured on the posteroanterior X-ray images, including global CB (C7 plumbline-central sacral vertical line,
The present study extends the knowledge on pain and gender providing data concerning the relationship be- tween computer usage, work ability, productivity, phys- ical activity,
tion to these generating functions, integral representations of the twisted q -Euler zeta function (and l -functions) are obtained, which interpolate the (generalized) twisted q -
Miyajima, “On the Hyers-Ulam stability of the Banach space-valued di ff erential equation y λy ,” Bulletin of the Korean Mathematical Society , vol. Takahasi,
Thus if process is to be sustained in the principal case, it must be on the basis of present business activity. The test which deter- mines whether or not a
Keywords: Emergence Christianity; Emergent Christianity; Great emergence; Phyllis Tickle; Christianity in the United States; Christianity in South Africa.. The Great Emergence:
Hierdie dienswerk moet ook telkens wanneer dit geïnstitusionaliseer word, in ‘n saaklike kontinuïteit met die historiese Jesus staan.. Die saak wat Jesus
