The Weighting is the Hardest Part: On the Behavior of the Likelihood Ratio Test and the Score Test Under a Data-Driven Weighting Scheme in Sequenced Samples
11
0
0
Full text
Figure
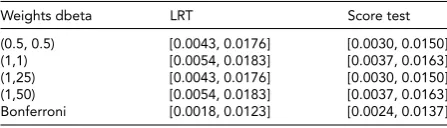
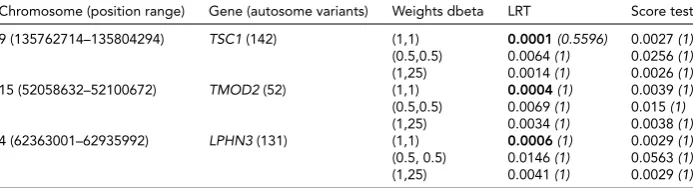
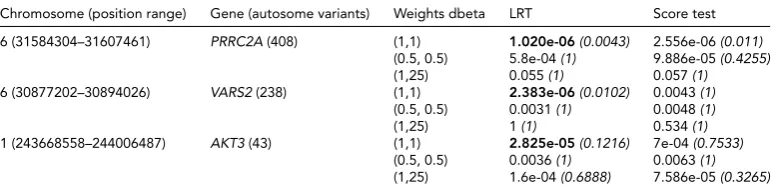
Related documents