Direct Transcriptional Consequences of Somatic Mutation in Breast Cancer
Full text
Figure
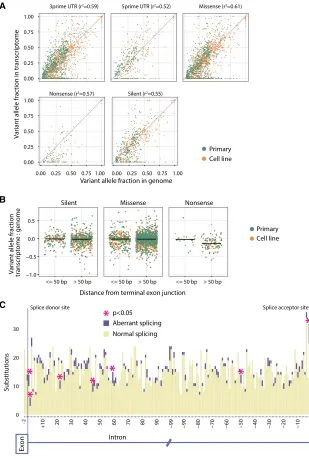
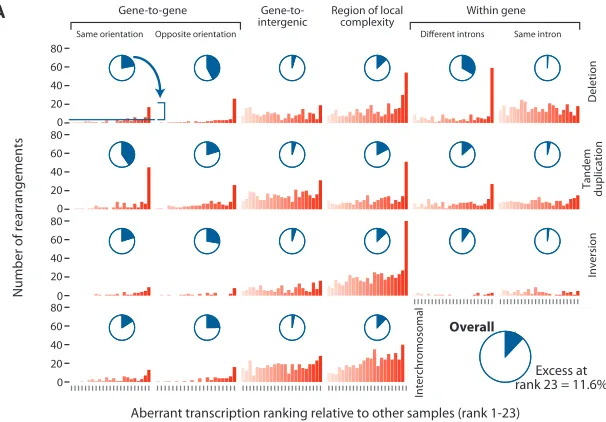
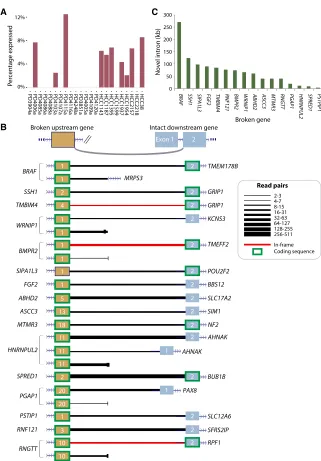

Related documents
The results of this part include the optical flow calculated from two sequential SST images and the geostrophic currents estimated by altimetry along-track data from multi-missions
To return to the notion of human rights language as a double-edged sword in the public health and human rights movement, some contend that the rhetorical power of
Thus, relation between profitability and dividend behaviour of the firm is supported with Catering and Signaling theory which in practice states that change in
European central Bank: Impact of sovereign wealth funds on global financial markets.. Occasional
(Moreover, in the U.S. banks meet reserve requirements on a lagged basis—required reserves for each two-week maintenance period are based on banks’ average deposits outstanding
machine 23 managed machines 23 no direct connection 28 trusted software 23 , 42 services network infrastructure 16 signed packages 46 software ClickOnce 20 packages 45 provisioning
Since, the nutraceuticals include dietary supplements, functional foods, etc., the components used in the formulation must be of food grade. This limits the choices for
Our study, despite limitations posed by small sample and its retrospective analisys, highlights the role of goiter ’ s extention (below the aortic arch), disease length (for more
