m
University College London
Phosphine Stabilized
Di-iron^iydfWe^
Complexes
Mark H. Lavender
Supervisor Dr Graeme Hogarth
1995
ProQuest Number: 10016768
All rights reserved
INFORMATION TO ALL USERS
The quality of this reproduction is dependent upon the quality of the copy submitted.
In the unlikely event that the author did not send a complete manuscript
and there are missing pages, these will be noted. Also, if material had to be removed, a note will indicate the deletion.
uest.
ProQuest 10016768
Published by ProQuest LLC(2016). Copyright of the Dissertation is held by the Author.
All rights reserved.
This work is protected against unauthorized copying under Title 17, United States Code. Microform Edition © ProQuest LLC.
ProQuest LLC
789 East Eisenhower Parkway P.O. Box 1346
This thesis describes the synthesis and reactivity of phosphine stabilised dimetailic complexes containing bridging-hydride ligands. The complex [Fe2(CO)6(fi-CO)(p.-dppm)] (dppm = PhgPCHgPPhg), reacts in the presence of uv irradiation with secondary phosphines to afford the complexes [Fe2(CO)4(jLi-CO)(|i-H)(|i-PR2)(p.-dppm)] (1.Ph, R=Ph; 1.Cy, R=Cy). These have been found to display two general types of reactivity, namely insertion of unsaturated organic molecules into the metal-hydride bond, and elimination of the hydride ligand with replacement by a 3-electron donor.
A variety of different organic molecules can be inserted affording a number of different products. Reaction with primary or activated secondary alkynes results in g-k vinyl complexes of the general form [Fe2(C O )4 (p.-CR'=CHR")(|i-PR2)()i-dppm)] (R" = H for 1° alkynes). Heterocumulenes such as carbon disulphide and isothiocyanates also readily insert into the metal-hydride moiety in 1 .Cy, to afford a dithioformato complex [Fe2(CO)4()i-S2CH )(|i-PCy2)(|i-dppm)] and formimidoyl, [Fe2(CO)4(|X2-R N=C H)(|i-PC y2)(|i-dppm)] and N-thioform am ido [Fe2(C O )4(|x-R'NCHS)(p.-PCy2)(|i-dppm }] com plexes respectively.
Contents
Chapter 1 General introduction
1 . 1 Carbonyl Ligands 13
1 . 2 Hydride Ligands 15
1 .2 . 1 Bonding in Hydride Ligands 15
1 .2 . 2 Synthesis of Hydride Ligands 17
1.2.3 Reactions of Hydride Ligands 19
1.2.4 Hydrides as Catalysts 2 1
1.3 Phosphorus-containing Ligands 2 2
1.3.1 Terminal-Phosphine 23
1.3.2 Terminal-Phosphido 24
1.3.3 Bridging-Phosphido 24
1.3.4 Diphosphines 29
1.3.5 Phosphorus-31 NMR Spectroscopy 32
1.4 Summary 35
1.5 References 36
Chapter 2 Synthesis and Structure of Hydride Complexes
[Fe2(CO)4(p-COKp-HXp-PRgKp-dppm)] (R = Ph, 1.Ph; R = Cy, 1.Cy)
2 . 1 Introduction 42
2 . 2 Synthesis of [Fe2(C0 )4(p-C0 )(|Li-H)(p-PR2)(p-dppm)] 44 2.3 Characterisation of [Fe2(C0 )4(p-C0 )(p-H)(p-PR2)(p-dppm)] 46
2.4 Possible Mechanism for Hydride Formation 52
2.5 Summary 53
2 . 6 References 55
Chapter 3 Reactions of [Fe2(CO)4(p-CO)(p-H)(p-PR2)(p-dppm)] with
Alkynes
3.3 Reaction with P ropy ne 73
3.4 Reaction with Phenylethyne 75
3.5 Selectivity of the Insertion Reactions of Alkynes with 1.Ph 82 3.6 Selectivity of the Insertion Reactions of Alkynes with 1.Cy 83 3.7 a-K Vinyl Fluxionality
3.7.1 Introduction 84
3.7.2 [Fe2(CO)4(p-CH=CH2)(p-PCy2)(|i“dppm)] 85
3.7.3 [Fe2(CO)4(p-M eC=CH2)(|i-PCy2)(p-dppm)] 87
3.7.4 [Fe2(CO)4(p-CH=CHMe)(^i-PCy2)(MPPm)] 87
3.7.5 [Fe2(CO)4(p-CPh=CH2)(p-PPh2)(ii-dppm)] 8 8
3.7.6 [Fe2(CO)4(|i-CH=CHPh)(^-PCy2)(M P P m )] 8 8
3.8 Reaction with Propargyl Alcohol 90
3.9 Proposed Mechanism for Reactions with Primary Alkynes 93
3.10 Reaction with Secondary Alkynes 94
3.11 Reaction of [Fe2(CO)4(|i-C O )(p-H )(|i-PPh2)(p.-dppm)] with
Dimethyl Acetylenedicarboxylate 97
3.12 Thermolysis of [Fe2(C0 )4(p-CR'=CHR')(p-PCy2)(p.-dppm)]
(R ’ = C0 2 Me) 1 0 0
3.13 Reaction of [Fe2(CO)4(p.-CO)(p-H)(ii-PCy2)(|i-dppm)] with
Dimethyl Acetylenedicarboxylate 108
3.14 UV Irradiation of [Fe2(CO)4(|i-C R ’=C H R ’)(|i-PCy2)(p-dppm)] 109
3.15 Summary 115
3.16 References 116
C hapter 4 R eactions of [Fe2(C O )4(p-C O )(p-H )(p-P R2)(|J-dppm)] with C um ulenes, H eterocum ulenes and Isonitriles
4.1 General Introduction 1 2 2
4.2 Reaction with Allene
4.2.1 Introduction 130
4.3.1 Introduction 134
4.3.2 Reactivity 136
4.4 Reaction with isothiocyanates
4.4.1 Introduction 137
4.4.2 Reactivity 137
4.5 Reaction with Nitriles and Isonitriles
4.5.1 Introduction 146
4.5.2 Reactivity 148
4.6 Reaction with Ethyl Diazoacetate
4.6.1 Introduction 150
4.6.2 Reactivity 151
4.7 Summary 153
4.8 References 154
Chapter 5 Elimination Reactions
5.1 General Introduction 157
5.2 Bis(phosphido) Bridged Complexes
5.2.1 Introduction 157
5.2.2 Synthesis 159
5.2.3 Proposed Mechanism for Formation 165
5.2.4 "Windshield-Wiper" Fluxionality 169
5.3 Thermolysis of [Fe2(CO )4(p-C O )(p-H )(|i-PR2)(ii-dppm)] and
Benzene Loss 173
5.4 Hydroxide Bridged Complexes
5.4.1 Introduction 179
5.4.2 Synthesis 183
5.5 Halide Bridged Complexes
5.5.1 Introduction 189
5.5.2 Synthesis 192
5.5.3 Proposed Mechanism for Formation 201
5.6 Summary 203
Aqueous Acids
6 . 1 General Introduction 209
6 . 2 Reaction with Hydrogen Halides 2 1 2
6.3 Reaction with Carboxylic Acids
6.3.1 Introduction 214
6.3.2 Reactivity 217
6.4 Reaction with Oxalic Acid
6.4.1 Introduction 224
6.4.2 Reactivity 224
6.5 Reactions with Other Acids 226
6 . 6 Proposed General Mechanism for Reactions with Acids 227
6.7 Summary 231
6 . 8 References 232
Concluding Remarks 234
Chapter 7 Expérimentai
7.1 General Comments 237
7.2 Experimental for Chapter 2 238
7.3 Experimental for Chapter 3 239
7.4 Experimental for Chapter 4 248
7.5 Experimental for Chapter 5 255
7.6 Experimental for Chapter 6 263
Appendices
Appendix 1 Crystal log raphic data
General Comments 272
1.3 [Fe2(C0 )4(p-CPh=CH2)(p-PPh2)(p-dppm)] 281
1.4 [Fe2(C0 ),(|i-C H =C HPh)(p-PPh2)(p-dppm)] 285
1.5 [Fe2(CO )4(p-CH=GHPh)(|x-PCy2)(ix-dppm)] 289
1 . 6 [Fe2(G0 ),{p-G(CH2 0 H)=CH2}(p-PPh2)(^i-dppm)] 293 1.7 [Fe2(G0 )4{|i-G(G0 2 Me)=GH(G0 2 M e)}(|i-PPh2)(|i-dppm)] 297 1 . 8 [Fe2(G0 )4{p-G(G0 2 Me)=GHG(0 M e)=0 }(|i-PPh2)(|i-dppm)] 301 1.9 [Fe2(GO)4{|i-GH2GHGH2NG(H)S}(p-PGy2)(p-dppm)] 305 1 . 1 0 [Fe2(GO)4(p-GH2GHGH2N=GH)(p-PGy2)(ii-dppm)] 309
1 . 1 1 [Fe2(G0 )4(p-PPh2)2(p-dppm)] 313
1 . 1 2 [Fe2(GO)4([i-PPh2)(|i-PPhGH2PPh2)] 317
1.13 [Fe2(GO)4(p-OH)(p-PPh2)(|x-dppm)] 321
1.14 [Fe2(GO)4(|i-GI)(p-PGy2)()i-dppm)] 325
1.15 [Fe2(GO)4(p-Br)(p-PGy2)(p-dppm)] 329
1.16 [Fe2(G0 )4(p-l)(p-PGy2)(|i-dppm)] 333
1.17 [Fe2(GQ)4(p-Ü2GH)(p-PGy2)(p-dppm)] 337
Appendix 2 Table of data for N-thioformamido and formimidoyl
complexes 341
Appendix 3 Table of pk^ values of selected acids 342
Appendix 4 Graphs of phosphide chem. shift vs. M-P-M bond angle 343
Appendix 5 Graph of M-P-M bond angle vs. M-M bond length 345
1 [Fe,{COU\i-H){[i-CO){[L-PR,){^-6ppm)]
2 [Fe2(CO),(^i-HC=CH2)(^i-PR2)(MPPm)] 3 [Fe2(CO)4([i-MeC=CH2)(^i-PR2)(MPPm)]
4 [Fe2(CO)4(|i-CH=CHMe)(n-PR2)(MPPm)]
5[Fe2(C0)Xp-PhC=CH2)(^i-PR2)(p-dppm)]
6 [Fe2(C0 ),(p-CH=CHPh)(p-PR2)(MPPm)] 7[Fe2(C 0)>-(C H 20H )C =C H 2}(^i-PR 2)(M PPm )]
8 [Fe2(C0)4{p-CH=CH(CH20H)}(^-PR2)(MPPm)]
9 c/s-[Fe2(CO),(p-CR'=CHR')(p-PR2)(p-dppm)]
1 0 frans-[Fe2(CO)4(ii-CR’=CHR’)(ii-PR2)(MPPm)] 11 [Fe2(CO)Jf|"-CR =CH-C(OMe)=0}(|i-PR2)(MPPm )]
12 [Fe2(CO),(p-S2CH)(p-PR2)(MPPm)]
13 [Fe2(CO),{|i-(CH2CHCH2)NCHS}(^i-PCy2)(MPPm)] 14 [Fe2(CO),{p-(CH2CHCH2)N=CH}(^-PCy2)(MPPm)]
15 [Fe2(CO),{|i-(CH3CH2)NCHS}(p-PCy2)(MPPm)] 16 [Fe2(CO)Jp-(CH3CH2)N=CH}(p-PCy2)(MPPm)] 17 [Fe2(CO),(p-PhNCHS)(^i-PCy2)(p-dppm)]
18 [Fe2(CO),(^i-’BuN=CH)(p-PR2)(Mppm)] '
19 [Fe2(C0),{p-HN-N==CH(C02Et)}(^i-PPh2)Mppm)]
2 0 [Fe2(C0 )/p-PR2)2(p-dppm)] 2 1 [Fe2(CO)5(^i-PR2)(p-PhPCH2PPh2)] 2 2 [Fe2(Ç0 )Xp-0 H)(p-PR2)(^-dppm)] 23 [Fe2(CO)Xp-CI)(^i-PR2)(p-dppm)]
24 [Fe2(C0)X^i-Br)(pi-PR2)(p-dppm)] _
25 [Fe2(C0)Xp-l)(n-PR2)Wppm)]
26 [Fe2(C0)/p-F)(|i-PR2)(p-dppm)}
27 [Fe2(C0),(p-02CH)(^i-PR2)(^i-dppm)]
28 [Fe2(C0)X|i-02CCF3)(|i-PR2)(|i-dppm)]
29 [Fe2(C0)/p.-02CCBr3)(^i-PR2)(p-dppm)]
30 [Fe2(C0),(K-02CCCl3)(^i-PR2)(|i-dppm)]
31 [Fe2(C0)X^i-02CCHCl2)(^-PR2)(p-dppm)]
32 [Fe2(C0),(p-02CCH2CI)(^i-PR2)(MPPm)]
33 [Fe2(C0),(p-02CC02H)(p-PCy2)(MPpm)]
34 [Fe2(CO)6(|i-PR2)(MPPm)r
R = Ph, Cy. R’ = COgM©
The abbreviation after the compound number corresponds to the substituent on the phosphide
Abbreviations
uv ultra-violet ir infra-red
nmr nuclear magnetic resonance
with reference to ir spectroscopy :
s strong
m medium
w weak
br broad
sh shoulder
cm'"' reciprocal centimetres
v(CO) carbonyl stretching frequency
with reference to nmr spectroscopy : ppm parts per million
s singlet
d doublet
t triplet
q quartet
qu quintet
m multiplet
dd doublet of doublets dt doublet of triplets
ddd doublet of doublet of doublets ddt doublet of doublet of triplets
br broad
Hz Hertz
J coupling constant Ô chemical shift
Dppm (Diphenylphosphino)methane Dpam (Diphenylarsino)methane Dmpm (Dimethylphosphino)methane Dppe (Diphenylphosphino)ethane □ C M dichloromethane
T H F Tetrahydrofuran
DMADDimethylacetylenedicarboxylate DEAD Diethylacetylenedicarboxylate
Me Methyl
Et Ethyl
'Pr /-Propyl "Bu /7-Butyl 'Bu f-Butyl
Ph Phenyl
Cy Cyclohexyl
Op Cyclopentadienyl
Op* Pentamethylcyclopentadienyl
py Pyridine
OTf Triflate Men Menthyl
dpp diphenylphosphido dcp dicyclohexylphosphido cod cyclooctadiene
pz pyrazolide
mpz 3-methylpyrazolide dmpz 3,5-dimethylpyrazolide bmpz 3(5)-bromo,5(3)-methyl
Except where acknowledged, all the work reported in this thesis was carried out by the Author in the Department of Chemistry, University College, London, between the October 1991 and the 31®* September 1994.
Acknowledgements
My sincere thanks to Dr. Graeme Hogarth for his constant guidance throughout this work. Both his depth of knowledge of bimetallic complexes and enthusiasm to further this area have been an inspiration.
I would also like to thank U.G.L. for financial support in the form of an Access fund during my three years of research and also a Graduate School award to enable myself to attend the XVI*^ International Conference of Organometallic Chemistry. I am also indebted to my grandmother who regularly helped me financially.
Thanks are also due to Dr. Andrea Sella for practical help in the laboratory and all the past and present members of labs 245 and 243 : Andy, Slobby, Carol, Donna, Emmanuel, Graham, Helen, Janella, Jim, Jo, Joe, Lai, Maya, Simon, Sung Ying, Tim, Tiz and Woo Sung for making the department a home away from home. I hope the endless hours playing Risk were as memorable to them as they are to me.
Some of the experiments with aqueous acids were performed by Miss Danya Corby and several of the insertion reactions by Mr. Khalid Shukri, both under the supervision of the Author. To these two, I am especially grateful for their hard work and dedication. Cyclic voltammetry of the bis-phosphido species, [Fe2(CO)4(|j,-PPh2)2(^-dppm)] was carried out by Miss Navjot Sahota. I am also indebted to Ms Jill Maxwell and Mr. Alan Stones for chemical analyses performed and to the departmental mechanical workshop for creating ’Pandora’s UV Box’ in which the photolytic reactions were carried out.
zoid rest in the shadoiu o f the Mmighty.
^ I unïï say o f the LOH^, 'He is my refuge and my fortress,
my Qod, in zuhom I trust."
^ Surely he zoild save you from the foiuhefs sruire and from iJie deadhy pestifence.
" He iviCC œver you iv ith his feathers,
and under his zvings you ziHdCfind refuge; his faithfuCness zûiCC he your shieCd and
rampart.
^ Jou zuid not fe a r the terror o f night, nor die arrozv tha t f des By day, ^ nor the pestdehce th a t staÛçs in the dar/qtess,
nor the pCague th a t destroys a t midday. ^ Si thousand may fadd a t your side,
ten thousand a t your rig h t hand, hut it iv id not come near you.
' ^ou zvid onSy observe zvith your eyes
and see the punishment o f the zviched. ^ I f you malçe the M ost H igh your dzueding
-even the L O I^ , zuho is my refuge - " then no harm zvid hefad you,
no disaster zvid come near your tent.
" SFor he zvid command his angeCs concerning
you
to guard you in a d your zvays; they zvid Cift you up in their hands, so tha t you zvid not strihe your fo o t against a
stone.
^ you zvid tread upon the [ion and the cohra; you zvid trampCe the great Cion and the
serpent.
"[Because he Coves me," says the LXO^RCp, "I zvid rescue him;
I zvid protect him, fo r he adqiozvCedges my name.
He zvid cad upon me, and I zvid anszver him; I zvid he zvith him in trouhCe,
I zvid dehver him and honour him. lAiith Cong Cife zvid I satisfy him
Chapter 1
General Introduction
This thesis describes the synthesis and reactivity of phosphine stabilised df-iron complexes, [Fe2(CO )4(|i-C O )(|i-H )(|i-P R2)(|x-dppm)] (R = Ph, Cy), containing a bridging-hydride ligand
8
(CO)2F e ^ -^ F e (C O) 2 H
Ph2P \ ^ P P h2 H2
It has been shown in recent years that organic molecules can be readily coordinated to dinuclear metal centres to form stable species. This opens up the opportunity for the synthesis of a rich variety of new complexes which may be useful in giving information on how organic molecules behave when attached to metal surfaces (heterogeneous catalysis), and which may provide new routes to forming unique coordination geometries and reactivity at the binuclear centre. During the course of this thesis the reader will encounter a variety of different metal-ligand interactions, the most common of which are briefly described below.
1.1 Carbonyl Ligands
The CO ligand can be coordinated in several ways to metal centres, either terminally (A) or bridging two or more metals (B-D). When bound to a single metal atom the ligand acts as a two-electron donor and is coordinated via a single o-bond. There are a number of ways in which a carbonyl ligand can bridge metal centres, most often observed being types (B) and (0).
o o
II V
c C
/ \ / \
1--- M M--- 1
(B) (C)
Ç c c c
M M M--- IVI M---M M---M
Chapter 1
Symmetrically bound (B) is most common with homometallic binuclear complexes where the geometry about each metal centre is the same, the ligand acting as a two-electron donor/ Type C is termed "semi-bridging" or asymmetric and is common for complexes in which the two metals have different coordination environments or oxidation states, for example in [Fe2(C O )6{C2(O H )M e)2], known as a "non-sawhorse" structure, the ligand acts to stabilise and balance the difference between the environments of the two iron atoms.^
....
OC'^|'®'^CO c o
The third type of bridging-carbonyl (D) is more rare. The ligand is formally a four-electron donor and linearly bridges the two metals as in [Mn2(CO)4(p.-fj\fi^-CO)(ii-dppm)].® This type of bonding is comparable to that of isonitriles bridging two metal centres (see Chapter 4).
Terminal and bridging carbonyls can be differentiated very easily on the basis of ir spectroscopy, the 'ü(CO) frequency being directly related to the 0 - 0 bond order. Hence, terminal carbonyls with a bond order of approximately three appear at a higher frequency (1800-2000cm'^) than bridging carbonyls (1700-1800cm ’'').
To induce reactivity in transition metal complexes containing such ligands often requires decarbonylation in order to produce a vacant coordination site. This can be effected either thermally or photochemically. Photolysis was chosen for the purposes of this work to minimise the number of starting reagents and to avoid thermal side-reactions which commonly cause phosphorus-carbon bond cleavage in complexes of the type studied.
The first reported use of ultra-violet irradiation to effect decarbonylation was in 1905" when Dewar and Jones prepared [Fe2(C0 )g] from [Fe(CO)J.
Since then, this process has been used to substitute many other ligands for carbonyls including olefins^ and phosphines.®’^
1.2 Hydride Ligands
Hydride ligands are also common in organometallic chemistry, and again can bind terminally or act as a bridge between two or three metal centres. There are examples of asymmetric hydride bridges indicating that the ligand is sensitive to the environment of the respective metal atoms.®
1.2.1 Bonding in Hydride Ligands
Terminally coordinated hydride ligands exhibit an average metal-hydride bond distance of 1.6 - 1.7Â consistent with a normal covalent linkage.® The bonding involved in bridging-hydride complexes has for a long time been a matter for debate. The three-centre, two-electron unit can be drawn in two ways, with (A) or without (B) metal-metal bonding.
M M M M
(A) (B) (C)
Comparisons of crystallographic data of complexes with and without hydride-ligands^® suggest only small changes in the M-M bond lengths upon coordination of the hydride. For example, comparing [Mo2Cp2(CO)4
(|j.-H)(|i-bond
PMe2)] with [Mo2Cp2(CO)g], only a slight lengthening of the metal-metal from 3.235(1 )Â to 3.267(2)Â is noted on hydride coordination. Also, the hydride complex [CpFeMn(C0 )g(|i-H)(|i-PPh2)] exhibits a bonding metal-metal distance of 2.806(1
)A
compared with the non-bonding interaction of 3.718(1)A
in CpFeMn(C0 )g(|i-PPh2) { M '- C (0 )M e}].''Chapter 1
[W2(C0 )g(N0 )(|i-H )]/^ the axial ligands do not point directly at the hydrogen atom suggesting bonding of the type formulated for boron hydrides of a closed three-centre bond, the three orbitals overlapping in a common region of space as shown in (C) and also below.
The first evidence for the existence of symmetrical three-centre, two-electron M-H-M bonds in transition metal complexes was given by Doedens and DahU^ who reported the X-ray crystal structure of [Mo2Cp2(CO )4 (|i-H)(|i-PMe2)]. They concluded that as the two metal atoms were in the same coordination environment, the bridging-hydrogen atom must be equally associated with each molybdenum atom (i.e. not bound directly to only one) and be situated equidistant from the two metals. However, due to the fact that the bridging-hydrogen atom usually lies on a crystallographic plane of symmetry, the latter observation may not be true but that the hydride is statistically symmetrical. In other words, it is time-averaged due to being randomly distributed in several equivalent sites displaced from the centre (although still associated with both metal atoms). A fundamental problem with most X-ray diffraction studies is that the scattering of X-rays is roughly proportional to the number of electrons in an atom, and so large metal atoms tend to ’dwarf’ smaller, nearby atoms, in particular hydrogen atoms, making their location difficult. This showed the limitations of using X-ray diffraction studies on these complexes, and so Dahl et al.^° investigated the same hydride species using neutron diffraction studies, the difference being that hydrogen atoms scatter neutrons with about the same efficiency as most other elements. Dahl showed that the hydrogen atom occupies a distinct coordination site and resides in a ’single-minimum potential well’, in other words, the hydrogen oscillates about a single equilibrium point equidistant from
the two metals, as opposed to randomly oscillating between two points displaced from the centre. This conclusion was drawn from the small thermal ellipsoid for the hydride atom compared with other hydrogen atoms in the crystal lattice. In the complex, [Mo2Cp2(CO)4(|i-H )(|i-P M e2)], the hydride ligand subtends an angle of 122.9(2)° to the two metal atoms from which it is equidistant, [M o(1)-H = 1.851(4)A; Mo(2 )-H = 1.869(4)] (a difference of 2.7a was considered to be just within experimental error).
Another way of determining the position of the hydride is to synthesise the related gold-phosphine complex, as in almost all clusters where structural comparisons are available, the "Au(PPh3)" unit occupies the same position as the isolobal hydride ligand in the related complex.""^
There are examples of complexes containing triply-bridging hydrides such as [HFeCo3(CO)g{P(OMe)3}3] in which the hydrogen caps the tricobalt face, being located 0.978(3)A from it. The M-H(average) bond length is 1.734(4)A and the angle the hydride ligand subtends, M-H-M is 91.8(2)°. Interstitial hydrogen atoms are also known, one of the first to be crystallographically observed was that of the anion [HC0 g(C0 ) i5]" in which the hydrogen atom is situated in the centre of the hexacobalt cage.^^
1.2.2 Synthesis of Hydride Complexes
Chapter 1
of the product is dependent on the type of metal atoms in the precursor and on the bridging ligands. For instance, changing the diphosphite ligand in the above example for the diphosphine dppm affords the analogous bridging-hydride species as does changing the metals to iron, hence affording [Fe2(C0 )4(|i-C0 )(|i-H)(|i-dppm )2][BFJ, the formation of which can be reversed by using a source of H~ such as sodium borohydride. The similar diruthenium complex [Ru2(C0 )4(p.-C0 )(p.-H)(|i-dmpm)2][BFJ^^ was prepared by the protonation of [Ru2(CO )4(|i-CO)(ji-dmpm)2] with HBF4.Et2 0 , and the same acid was used in the formation of the dimolybdenum species [Mo2Cp2(CO)4(|i-H )(|i-dppm)],^® and the mixed-metal complex [RhRe(CO)4(|i-H)(|i-dppm )2][BF4].‘'®
Di-iron nonacarbonyl is not readily protonated. It reacts with hydrogen halides under uv irradiation, but rather than give protonated derivatives, it affords bridging-halide species [Fe2(C0)6(|i-X)2].^° Replacement of two carbonyls by stronger electron-donating phosphine ligands increases the basicity of the di-iron centre, thus facilitating protonation. Hence reaction of [Fe2(C O )6(ii-CO)(|i-dppm)] with HBF4.Et2 0 yields the hydride species [Fe2(C O )6(ii-CO)(|i-H)(^-dppm )][BF4].^^
More stable, neutral hydride complexes can be synthesised by protonation of anionic binuclear complexes.^^ Thus, reaction of [NEt4][Fe2(CO)7(|i-PPh2)] with trifluoroacetic acid in TH F affords the neutral species [Fe2(C O )7(|i-H)(|i-PPh2)] characterised as a hydride by the low field resonance at -610.0 in the ^H nmr spectrum.
Bimetallic complexes containing both terminal and bridging-hydride moieties are known. The mixed-metal species [RuRhH2CI(f|^-C0D)(fi^-dppm)(|i-dppm)] formed by the combination of [RuH2(dppm)2] and [RhCI(C0D)]2 contains both types of hydride, each of which gives rise to a low field resonance in the ^H nmr spectrum.^^ This complex is also of interest as it contains dppm ligands in both chelating and bridging forms, (see later section).
'H nmr (CsD^) h p
P \* ^ '/P H2 = -515.6 (J = 22Hz)
H., = -514.6 (J = 21 Hz)
Bridging-hydride complexes may also be produced from the direct interaction of transition metal complexes with molecular hydrogen. However, oxidative-addition of dihydrogen may promote subsequent cleavage in bridging-diphosphine ligands. Thus, bubbling Hg through a solution of [Ru3(C0)g(dppm)g] at 85°C affords [Ru3(CO)6(|i-H)2(|i-PPhC H2PPh2)2]^'^ with loss of carbon monoxide and benzene (see chapter 5). The analogous reaction
bis
containing dpamjdiphenylarsenomethane) has also been performed, in this case an intermediate in the reaction was isolated and identified as the mono hydride species [Ru3(CO)7(|LL-H)(|i-AsPhCH2AsPh2)(p-dpam)]. Refluxing [Ru3(C0 )gL2] (L=dppm, dpam) does not give rise to any cleavage products and so the oxidative-addition of dihydrogen almost certainly precedes the cleavage of the ligand.
A more recent preparation, involving chloride substitution, was reported by Riera et al.^^ Thus, the reaction of Li[HBEt3] with the dichloride [Mn2(C0 )e(|i-Cl)2(|i-dppm)] at room temperature in THF after aqueous workup affords the 32-electron dihydride [IVIn2(C0 )g(|i-H)2(|i-dppm)]. This is, however, not a simple substitution reaction of Cl" for H" as it is dependent upon the number of equivalents of Li[HBEt3] added, two for each chloride replaced. Secondly, use of Li[DBEt3] and DgO led to the formation of [Mn2(CO)6(|i-D)(ji-H)(}i-dppm)] suggesting that the source of the second hydride in the non-deuterated reaction is different to the first. Therefore these types of reactions have been shown to be more complex than at first appears.
1.2.3 Reactions of Hydride Complexes
Chapter 1
continues to attract attention due to the critical importance of such species in many stoichiometric and catalytic reactions. For polyhydrides, this stems from an ability to lose hydrogen by a variety of means. The coordinatively unsaturated intermediates generated may react with donor molecules to give new complexes, or function as homogeneous catalysts in reactions with various organic molecules.
Polyhydrides often undergo reactions with donor molecules, which replace Hg in the complex, reactions which can sometimes be reversed by the action of Hg:
L'
L|-|MH2 —- LriM“L'
Hz
The photochemistry of polyhydrides has great potential in opening active sites in complexes which are otherwise thermally stable. These reactions depend on the photoextrusion of Hg from the complex, which appears to be concerted, and does not involve radical or ionic intermediates.^® The reaction of the triosmium dihydride [OSg(CO)io(|i-H)g] with ethyne leads to formation of the o,E-vinyl complex [Os3(CO)io(M--H)(ji-CH=CHg)] at room temperature, the latter being converted to [Os3i(CO)g,(p-H)g(|i-C=CHg)] under forcing conditions, in refluxing hexane. The unsaturated dihydride also reacts with methyl-substituted alkynes to give similar a,E-vinyl bridged complexes.^^ The ease with which this reaction takes place is almost certainly associated with the ability of [Os3(CO)io(p-H)g] to coordinate an additional two-electron donor ligand.^®’^®
Os(CO)4 Os(CO)4 ^ ^ C H g
(C0 ),0 ^ ^ 0 . (0 0 ) . Ü S = ^ ( C O » i ü i o . ( C O | . ^ £ 5 - » (0 0 ).0 . Æ
The reaction of coordinatively saturated complexes containing bridging-hydride ligands often requires more forcing conditions. Thus, tetranuclear [Os4(C O )i2()i-H)J reacts with ethyne under photolytic conditions to give [Os4(C O )tt(|i-H )3(|i-C H=C H2)],^°’®^ but other complexes studied appear not to give simple insertion products. Thus, the photolytic reaction of [Re3(C O )i2 (p-1-1)3] with diphenylacetylene gives dimeric substitution products [Re2(C0 )7 (p-H)2(|i-PhC=CPh)2] and [Re2(C0 )5(p-H)2(|i-PhC=CPh)3],^^ while the reaction of [FeRu3(C O )i3(ii-H)2] with RC=CR^ gives [FeRu3(C O )i2(p-RC=CR^)] with elimination of molecular hydrogen.^^ The reaction of [IV1n2(C0 )g(p-H)(p-PPh2)] with RC=CR^ does, however, give simple products of the general form [Mn2(CO)7(ji-PPh2)(|i-CR=CHR^)f'^ (R, R^ = H, Ph, CF3) derived from the parent complex by insertion of the alkyne into a manganese-hydride bond.
/ \ ’ 7 \ ’
/ \ RP=PR ' / \
(CO)4Mn Mn(CO)4 — — ( CO)4Mn Mn(CO)3
„_v/
^C H R
1.2.4 Hydrides as Catalysts
In reactions of polyhydrides with alkenes, hydride ligands are almost always transferred to a second alkene molecule (Equation 1 ). Hydrogenation of the alkene complex can regenerate another hydride species and release a second alkane molecule (Equation 2). Thus polyhydrides can function as homogeneous catalysts for the hydrogenation or isomérisation of alkenes.
Equation 1, LnM H2 + 2 ^ --- ► LnM— 1| +
Equation 2, LnM— + 2 Hz --- ► LnMHz +
Chapter 1
= P(CH2CH2PPh2)3) and its nitrogen derivative.^^ It is thought that the dihydrogen remains coordinated to iron during the insertion reaction and the free coordination site for the activation of the primary alkyne is created by a temporary ’unfastening’ of one phosphine arm of the PP3 ligand. The reaction affords a o-vinyl complex which consequently loses an alkene molecule due to its instability containing an acidic f|^-H2 ligand and a basic vinyl moiety, cis to one another.
/ \
Fe H
H- -R
H e
“ w "
fe ^ H H
H R
Fe> = < '
\ H
He
H \
Fe
C = C / R
H
H H
Fe— H
H
Fe
R / H
H
Fe— C = C — R
The similar osmium complex [Os(PPg)(H)(N2)][BPhJ acts as a catalyst to the regio- and stereoselective dimérisation of phenylacetylene into (Z)-1,4-diphenylbut-3-en-1 -yne,^® a key step of which is the formation of the vinylidene species, [Os(PP3)(H){C=C(H)Ph}][BPhJ.
1.3 Phosphorus-Containing Ligands
Ligands containing phosphorus have been used extensively in the
chemistry of transition metals over recent years, due to the fact that such complexes are catalysts for a number of processes^^’^®'^® including hydrogenation, hydroformylation and polymerisation. Wilkinson and coworkers synthesised a variety of catalytically active triphenylphosphine complexes of ruthenium, for example, [RuHCI(PPh3)3] which catalyse hydrogenation and olefin polymerisation; however the best known example of such a catalyst is the rhodium complex, "Wilkinson’s catalyst", [RhCI(PPh3)3]. In order to stabilise the [Co2(CO)g] catalyst used in hydroformylation processes, ligand ’modifiers’ are added. In the 1960’s Slaugh and Mullineaux pioneered the use of tertiary phosphines as catalyst modifiers.
Such ligands appear to be extremely versatile in their ability to stabilise a wide variety of metals in a wide variety of formal oxidation states. This versatility stems from the ability of phosphorus ligands to vary their steric and electronic properties. Hence, simply by changing substituents on the ligand the reaction rate, selectivity and stability can be altered.
1.3.1 Terminal Phosphine, M-PR3
This is a simple phosphine derivative. The ligand acts as a two-electron donor due to the availability of the lone pair on the phosphorus atom. Transition metal complexes of this type are very common as they are very simple to synthesise under mild conditions. Prim ary,secondary"^^ and te rtia ry p h o s p h in e ligands are all known, usually synthesised by simple substitution reactions. For example, the mixed-ligand complex [Os(CO)CIH(PPhH2)(PPh3)2]^ was prepared by substitution of a bulky, labile PPh3 ligand."'
M PR3 M--- PR2 M =fc=P R 2
(A) (B)
Chapter 1
1.3.2 Terminal Phosphide, M-PRg
Unlike phosphines, terminal phosphide ligands can exhibit bonding with or without the phosphorus lone pair. Containing one less substituent, the phosphorus atom can bind in a a-fashion thus donating one electron (A), or can facilitate the lone pair and donate three electrons to the metal-phosphorus bond (B). This difference in bonding affects the geometry at the phosphorus atom which can be planar or pyramidal.
pyramidal
PPhg
eg:
R
(1 - electron
LnM" y
donor)
R
PPhs
0 = 106 -1140
planar
P
Cl. PEt^ph
( 3 -electro n
M e ,c c = v ^ p (
donor)
\ 8
.R
EtaP Cl
H
0 = 127- 1400
A comparison of the crystal structure of [OsCI(GO)2(PPh3)2(PPhH)]'^'‘ with that of [W (=GCM e3)(PHPh)Gl2(PEt3)2f^ clearly shows the distinctions. The metal-phosphorus-substituent bond angle ranges from 106-114° for pyramidal phosphorus complexes; and 127-140° for planar examples. There is also a difference in the metal-phosphorus bond lengths for the phosphide ligands and the tertiary phosphine ligands in the respective complexes. In the pyramidal case, Os-P is approx. 0 .1Â longer for the phosphide ligand compared with the phosphine, whereas for the planar complex W -P is approx. 0.26Â shorter for the phosphide ligand.
1.3.3 Bridging-Phosphido, P2-PR2
In complexes with two metal centres, the phosphorus-containing ligands
are able to bridge the two atoms whilst terminal coordination is still possible. These ligands are commonly used in binuclear complex chemistry as they are known to inhibit fragmentation to mononuclear species and they are relatively inert, although recent reports suggest that the latter is not always true (see later). Bridging-phosphido ligands are formally three-electron donors and subtend a wide-variety of angles to metal centres from between 70° for [Fe2(CO)6(ii-PPh2)(|i-CI)]"® and 107° for [CplVlnFe(C0)#-PPh2)(^i-C(0)IVIe}]."" This flexibility of the 1 1-PR2 moiety was observed by Carty"^® who noticed a direct relationship between ®^P nmr chemical shifts and the M -P-M angle (see following section and appendices). The versatility of this ligand is also illustrated in the possibility of fluxionality in bis(phosphido) complexes (Chapter 5). Complexes containing bridging-phosphido ligands are prepared in a number of different ways. Oxidative-addition of [M(CO)5(PPh2H)] (M = Cr, Mo, W) to [Pt(C2HJ(PPhg)2] yields the heterobimetallic species [MPtH(C0)5()i-PPh2)(PPh3)2] containing a terminal hydride which, under mild conditions, loses CO to afford [MPt(CO)4(|j.-H)(ji-PPh2)(PPh3)2]. The reaction is thought to proceed via PPh3 dissociation and a CO transfer via a bridging carbonyl intermediate. The result is sensitive to the acidity of the P-H and so replacing diphenylphosphine with dicyclohexylphosphine in [M(C0 )s(PR2H)] results in a less acidic P-H bond and hence a much slower oxidative addition step."^®
The first reported halide-substituted phosphide bridged complexes were prepared by reaction of the THF-solvated metal carbonyl species M (C 0)5.THF (M=Cr,W) with the mononuclear phosphide complex [CpCr(C0)3PCl2]/^ Oxidative-addition of phosphorus-carbon bonds also affords phosphide-bridged complexes. Thus, pyrolysis of the complexes [M3(C0)ii(Ph2PC=CR)] (M = Fe, Ru, Os; R = Ph, 'Bu, 'Pr), formed by the action of PhgPC^CR on [M3(C0)i2l in the presence of Me3N0 , initiates metal-metal bond cleavage and oxidative addition of the P-C (acetylide) bond across a metal-metal edge to afford [M2(C0)3(K2-f|'-C=CR)(^i-PPh2)].'°
Chapter 1
coordinated diphosphine,®^ and thermal cleavage of a vinylphosphine,^^ but perhaps the most common preparation is the simple oxidative-addition of a primary or secondary phosphine to a metal carbonyl complex. Hence, [Os3(CO)8(p-H)2(p-P'Bü2)2]^'^ is formed by refluxing [Os3(C O )i2] with 'BUgPH in THF. Interestingly, if uv irradiation rather than heat is used, [Os3(CO)ioCBu2PH)2] is the product, whereas prolonged heating of any of the above in di-n-butylether (143°C) affords [Os3(CO)7('Bu2PH)2(|i-H)2(|i3-*BuP)] containing a phosphinidine ligand.
Some results, however, suggest that metal-bound phosphido moieties are not as stable as initially expected and the ligands are sometimes non innocent. For example, the cluster [Ru3(C0)8(p-Ph2P-py)(p-PPh2)]^^ reacts with molecular hydrogen to afford [Ru3(C0 )g(p-H)(|i-Ph2P-py)(PPh2H)] in which the bridging phosphido ligand is converted to a terminal secondary phosphine.
A similar transformation occurs in the reactions of [Mo2(CO)8(p-PPh2)2] with BR3H" and UR nucleophiles in which cleavage of ^-(p-PPhg) bonds is proton induced.^® This has also been observed for [Fe2(C0 )6(p-PPh2)2].^^
Fe(CO)3
(CO UFe LiBEtgH
Ph z P
( C 0 ) 3 F e Fe(CO)2 PPhgH
Protonation of the dipalladium complex [Pd2(p-PR2)2(PH R2)2f® (R = 'Bu) with CF3SO3H or HBF^.EtgO yields a dimeric complex containing a secondary phosphine ligand with an ’agostic’ interaction involving an M...H-P fragment.
/ \ H—
/ \ CF3SO3H / / \
R2HP— Pd— - P d — PHR2 --- ►--- RgHP— P d - --- Pd— PHRg orHBF^.EtgO
R 2 R 2
R = tBu
The nmr spectrum showed four resonances corresponding to the four inequivalent phosphorus nuclei at 5455.3, 217.2, 52.0 and 47.7, and a hydridic proton signal was noted at -50.16 (coupled very strongly to one of the phosphorus atoms) in the nmr spectrum. Normally, protonation of such s p e c ie s a f f o r d s h y d r i d e c o m p l e x e s such a s [ W ( C0 )5 (|i-PPh2)O s(H )(C O )2(PMe Ph2)(PPh2H)]^® formed from the reaction of the mixed-metal species Li[W(CO)4(p-PPh2)2 0 s(CHO)(CO)2(PMe Ph2)], although even in this example a bridging-phosphido ligand is converted into a terminal phosphine. The phosphido ligand was reformed upon refluxing the hydride complex in toluene to afford the bis-phosphido complex, [W (C0 )4 (|i-PPh2)2 0 s(CO )2(PM ePh2)].
I
^
(C0)4W Os(CO)2(PMePh2) ■■ ^^^COOH ^ ( c o ) s W ^ ^ 6 s C ^ °
X
f= 0 HPh2p /H
heat
toluene
P h 2
(CO)4W^^Os(CO)2(PMePh2)
P h z
metal-Chapter 1
phosphorus bond.
2' '2
/ ' \ C,H
( C O ) ^ M n ( ' M n ( C 0 ) 4 —
P CH2CI2
^Ph (C0 )4Mn
Ph Ph
M n ( C 0 ) 2
PPhr
(CO>2Mh— -Mn(CO) 3
H HC
Similarly, uv irradiation of [Mn2(CO)a(|i-PPh2)2] with allene lead to the formation of
15
products, two of which were characterised as [Mn2(CO)7(|i-PPh2)(|LL-Ti^-C3H4PPh2)] and [M n3(C0),(n-PPh3)(M '-C6H,PPh2)].(C0 )4M n(^ ^M n(C0 > 4 R2
allene
CH2CI2
Ph2P
(C0 )4M n ^ ^Mn(CO) 3
P i / "Ph
CH2 ÇHg
H
(C0 )3Mn
y
--Ph Ph
1.3.4 Diphosphines, RgP-X-PRg
Diphosphines consist of two phosphine units joined by a linking atom or group of atoms. Most common linking groups are methylene, oxygen and amine, although a huge variety of others is known. There are a number of ways in which diphosphines can coordinate to one or more metal centres.
Chelation - The commonest example of a diphosphine chelating to a single metal centre is that of dppe, bis(diphenylphosphino)ethane. Here both phosphorus atoms coordinate to the metal centre via their respective lone pairs forming a 5-membered ring.
PPho
\
y C H ; P h z P ^ /Bis(diphenylphosphino)methane, dppm, is the diphosphine with methylene as the linking group. When dppm acts as a chelating ligand the diphosphine core and metal form a planar unit in which there is considerable ring strain. This is shown in the example®® of fra/7s-[RhHCI(dppm)2r where the phosphorus-metal-phosphorus angles are as acute as 67° and the angle subtended by the methylene carbon to the two phosphorus atoms is -9 5 °. Due to this strain there are few reported complexes containing chelating dppm®^ as compared with the ligand acting in a monodentate or bridging fashion.
Chapter 1
Monodentate - If the diphosphine ligand is bound to a metal centre by only one of its phosphorus atoms then it is termed monodentate. This occurs where the linking group between the two phosphorus atoms is either too bulky, or too long, or short to form a stable ring as above. The ideal ring size is five, any smaller increases the ring strain and favours the open, single coordinated structure. Dppm forms complexes such as this, for example [Fe(CO)Xf|^-dppm)] is thought to be an intermediate in the preparation of [Fe2(CO)6 (|i-CO)(|i-dppm)] (see Chapter 2).
Fe(CO)4
The most recent example of dppm binding to a bimetallic unit in this fashion is the di-iron complex [Fe2(C O )5(p,-P‘Bu2)(ii-PCy2)(i*|^-dppm)],®® formed by addition of dppm to the coordinatively unsaturated complex [Pe2(C0 )5 (|i-P'Bu2)(p-PCy2)]. As a result of the f|^-bonding, the two diphosphine phosphorus atoms exhibit quite different chemical shifts in the nmr spectrum at Ô62.7 (dd, J = 46, 12Hz) and -525.4 (d, J = 46Hz), the latter corresponding to the unbound phosphorus.
‘ BuP. .PCyg 'B u P . .PCyg
OC,, yC O dppm ^ OC,, .,CO
CO CO P h2P^ /P P h j
Bidentate - Monodentate complexes tend to be less stable compared with other dppm complexes as the free phosphorus atom is often oxidised to the phosphine oxide®^ or used to bind to another metal atom. If this is the case the diphosphine is acting as a 4-electron donor and bridges two metal atoms. This is the most common form of binding for dppm as it forms a stable 5-membered ring, the optimum ring size for such a system.
Puddephatt®^ argued that the role of the phosphine in such ligands is "to prevent dissociation of dimer to monomer, to promote bridging by other ligands and to promote binuclear reactions involving formation and cleavage of metal-metal bonds." He also showed how the steric bulk of substituents on the diphosphine considerably influence the reactivity. For example, oxidative-addition of methyl iodide and iodine readily occur for the complex [Me2 Pt(ji-dmpm)2PtMe2], however, the dppm analogue fails to react with methyl iodide and iodine causes cleavage of a methyl-platinum bond.®®
Along with the monodentate example above, Bottcher et al.®® have synthesised a di-iron complex bridged by dppm namely, [Fe2(C O )3(|i-P^Bu2 )(jJ.-PCy2)(p-dppm)]. In contrast to the monodentate species which contains an approximately planar Fe2P2 core (0 = 178.5°), this complex contains a puckered core (0 = 107.6°) caused by the steric requirements of the bridging-dppm ligand (see Chapter 5).
Shaw published a number of papers in the 1980s describing heterobimetallic complexes bridged by a dppm ligand. For example, reaction of the monodentate-dppm complex [Fe(C0 )4(f|^-dppm)] with [Rh2Cl2(CO)J affords the mixed-metal species [Fe(GO)4(p-dppm)RhGl(CO)].^°
CO ? ° ? '
o c - L < ! ° tR h .c i.(C 0 ).i ^
I CO I ^ c o I
P h 2 P ^ ^ P P ^ 2 PhzP^^^^PPhz
Chapter 1
I PhzP PPhz
I PhzPv..^^^PPh2
A similar method was used to prepare a number of other heterobimetallic species including [Ir-Mn]/^ [Cr,Mo,W -Fef^ and [Cr,Mo,W -lr,Rhf^ complexes. Complexes are known which contain three bridging p,-dppm ligands, for example [Pt2(|i-dppm)J^^ which adopts the 'manxane' structure. These types of complexes have been studied in depth and have a rich and varied chemistry.
PhzP;^ Pj^P P fh z^ P P h j
1.3.5 Phosphorus-31 NMR Spectroscopy
The stable isotope of phosphorus has a spin of l=V2. Although the phosphorus nucleus has one characteristic resonance frequency for a specified external field strength, shielding effects arising from chemical bonding, which influence the actual field strength at the nucleus, cause changes in the resonance frequency giving rise to ’chemical shifts’. These are usually measured with respect to the single sharp absorption in 85% orthophosphoric acid in water which is used as an external standard. A number of other standards can be used such as P(0 Me) 3 in organic solvents. There are two main factors which affect the chemical shift. These are electronegativity and the degree of jc-back bonding, both of which directly affect the shielding effects on the nucleus. If only the electronegativity of the substituent groups is allowed to alter, an increase in electronegativity, decreases the electron density on the phosphorus atom, causing deshielding, for example
PCI3 MePCl2 Me^PCl W\eJP
+219 +191 +94 -64
The relative intensities of resonances are proportional to the relative numbers of nuclei producing them. Comparatively large samples are, however, needed to study phosphorus resonances because of the rather low sensitivity of the element to magnetic fields. The relative sensitivity for equal numbers of atomic nuclei at constant field strength is
'H =1.00; '^0=1.59x10'^; '®F=0.833; ^'P =6.63x10^
Trivalent phosphorus nuclei are usually less shielded than pentavalent nuclei and show a spread of shifts ~500ppm whereas the latter have a spread of -lOOppm . A significant difference in chemical shift is seen when comparing different types of phosphorus ligands attached to transition metals. Terminal (pentavalent) phosphine ligands are seen to have similar values to those in Table 1.1 (Ô48.5 for the PEt^Ph ligand below).^®
Table 1 . 1 Some typical chemical shifts, ô(ppm) for phosphorus compounds
PH3 -240 PPhs -89
PlVte^ -64 PF
5 -80
PPh3 - 6 PH
2F3 -24
H3PO , 0 (EtO)3PO - 1
P(0 Et) 3 +137 POCI3 +3
PCI3 +219 Ph3P=CH2 + 2 0
Chapter 1
PPhz
EtjPhP,,, / \
,,C0
^ C o C o ^
“ =
V i
V
617 3 .4In complexes where there is no metal-metal interaction, an upfield shift to around 650 to -5200 is observed.
( C O ) 3 C o C o ( C 0 ) 3
/
-6 1 1 4 .2
This relationship between chemical shifts and metal-metal interactions has been investigated by Carty^^ amongst others. He showed that the chemical shifts for phosphorus nuclei bridging two metal centres are shifted downfield with a decrease in the metal-metal bond length and hence a closing of the M-P-M bond angle. For example, on increasing the bond order from 1 to 2 on going from [Fe2(CO)6(|i-PPh2)(|i-P ’Bu2)] to [Fe2(C O )5(p-PPh2 )(p-P‘Bu2)] (a decrease in bond length of -0 .2 4 Â ) causes a downfield shift from Ô294.4 to 6354.8 for the P*Bu2 group.
Coupling constants from nmr spectra can give valuable information about the stereochemistry of complexes. There is thought to be a direct relationship between the coupling constant between two phosphorus nuclei (Jpp) and the angle subtended by them (0).^® The relationship can be shown by a graph of Jpp plotted against 0 and can be understood in terms of orbital overlap. When two nuclei are in a trans disposition to one another, the angle subtended by them is approximately 180° and so almost maximum overlap of the bonding orbitals is possible. If, however, the two nuclei subtend to a much smaller angle, and hence to a cis configuration, the bonding orbitals have much less overlap consequently giving rise to a smaller coupling constant.
v i p p
(Hz)
e n 180
100
It is therefore possible to obtain a coupling constant of zero between two nuclei. This is true for the example below, where is seemingly only coupled to Pg and not to P^, the bridging phosphido. At a closer inspection, it is seen that the angle subtended by P^ and P^ is approximately 1 0 0 ° hence giving rise to a coupling constant of OHz.^®
ô5.9(^J = 1 8 .1 H z ^
Ù = 11.8Hz) Phz
_Ph
5 54.3 ("j = 18.1Hz)"
Ph2Pbv (CO^Fe
Ph:
Fe(CO)3
Ô 112.6 (‘j = 11.8Hz)
1.4 Summary
Chapter 1
although as shown (and investigated further in Chapter 5), phosphorus-carbon cleavage can occur under certain conditions.
The importance of the use of nmr spectroscopy in characterising new species has been stressed, especially the coupling constants between phosphorus nuclei which have been used extensively in this work to give information on symmetry within molecules and often clues to the presence of fluxionality within the system (Chapters 3 and 5).
Within each of the following chapters a more detailed introduction into other common ligands of such species will be given, in particular, those which are a result of the insertion of organic molecules into metal-hydride bonds (Chapters 3 and 4) and those formed by elimination of the hydride ligand (Chapter 5). Chapter 2 describes the formation of the hydride complexes [Fe2(C0 X(p-H)(p-C0 )(p-PR2)(p-dppm)] (R = Ph, Cy) and it is on the chemistry of these two species that most of this thesis is concerned. The final chapter (6 ) describes the reactions of the bridging-vinyl complex [Fe2(CO)4(|i-a,7 i-C H =i-C H2)(p-PCy2)(p-dppm)] (prepared in Chapter 3) towards various aqueous acids and the formation of new bridging ligands by the loss of ethylene.
1.5 References
1. E. G. Mednikov, N. K. Eremenko, S. P. Gubin, Yu. L. Slovokhotov and Yu. T. Struchkov, J. Organomet. Chem., 1982, 239, 401.
2. F. A. Cotton and J. M. Troup, J. Am. Chem. Soc., 1974, 96, 1233.
3. R. Colton, C. J. Commons and B. F. Hoskins, J. Chem. Soc., Chem. Commun., 1975, 363.
4. J. Dewar and H. O. Jones, Proc. Roy. Soc. (London) Ser. A , 1905, 76, 558. 5. W. Strohmeier, Angew. Chem., 1964, 76, 873.
6. R. S. Nyholm, S. S. Sandhu and M. H. B. Stiddard, J. Chem. Soc. 1963, 5916. 7. J. Lewis, R. S. Nyholm, S. S. Sandhu and M. H. B. Stiddard, J. Chem. Soc., 1964, 2825.
8. R. Bender, P. Braunstein, Y. Dusausoy and J. Protas, Angew. Chem. Int. Ed. E ngl., 1978, 1 7 , 596.
9. S. J. LaPiaca, W. C. Hamilton, J. A. Ibers and A. Davison, In o rg . Chem., 1969, 8, 1928; S. C. Abrahamson, A. P. Ginsberg and K. Knox, In o rg . Chem., 1964, 3, 558.
10. J. L. Petersen, L. F. Dahl and J. M. W illiams, J. Am. Chem. Soc., 1974, 9 6 , 6610. 11. R. Rosen, J. B. Hoke, R. R. W hittle, G. L. Geoffroy, J. P. Hutchinson and J. A. Zubieta, O rganom etallics, 1984, 3, 846.
12. J. P. Olsen, T. F. Koetzle, S. W. Kirtley, M . Andrews, D. L. Tipton and R. Ban, J. Am. Chem. Soc,. 1974, 9 6 , 6621.
13. R. J. Doedens and L. F. Dahl, J. Am. Chem. Soc., 1965, 8 7 , 2576. 14. I.D Slater, Adv. Organomet. Chem., 1989, 2 9 , 249.
15. D. W. Hart, R. G. Teller, C. Y. Wei, R. Bau, G. Longoni, S. Campanella, P. Chini and T. F. Koetzle, Angew. Chem. Int. Ed. E n g l, 1979, 1 8 , 80.
16. J. S. Field, R. J. Haynes, C. N. Sampson, J. Sundermeyer and K. G. Moodley, J. O rganomet. Chem., 1987, 3 2 2 , C l.
17. K. A. Johnson and W. L. Gladfelter, O rganom etallics, 1989, 8, 2866. 18. V. Riera, M. A. Ruiz and F. Villefane, O rganom etallics, 1992, 1 1 , 2854. 19. D. M . Antonelli and M. Cowie, In o rg . Chem., 1990, 2 9 , 4039.
20. E. Koerner von Gustorf, J. C. Hogan and R. Wagner, Z. N a turfo rsch , T e il B, 1972, 2 7 , 140.
21. J. Boothman and G. Hogarth, J. O rganomet. Chem., 1992, 4 3 7 , 201.
22. B. Walther, H. Hartung, H.-C. Bottcher, U. Baumeister, U. Bohland, J. Reinhold, J. Sieler, J. Ladriere and H. M. Schiebel, P olyhedron, 1991, 1 0 , 2423.
23. B.Delavaux, B. Chaudret, F. Dahan and R. Poilblanc, O rg an o m e ta llics, 1985, 4, 935.
24. G. Lavigne, O rganom etallics, 1982, 1 , 1041.
Tiripicchio 25. F. J. G. Alonso, V. Riera, M. A. Ruiz, A. Tiripicchio and M . Camellini, O rg a n o m e ta llic s , 1992, 1 1 , 370.
Chapter 1
27. A. J. Deeming, S. Hasso and M. Underhill, J. Organom et, Chem. 1974, 8 0 , C53. 28. A. J. Deeming and S. Hasso, J, O rganomet. Chem., 1975, 8 8 , C21.
29. J. B. Keister and J. R. Shapley, J. Organomet. Chem., 1975, 8 5 , C29. 30. J. Lewis and B. F. Johnson, Gazz. Chim. Ita l., 1979, 1 0 9 , 271.
31. J. W. Kelland, PhD Thesis, University o f Cambridge, 1979.
32. R. A. Epstein, T. R. Gaffney, G. L. Geoffroy, W. L. Gladfelter and R. S. Henderson, J. Am. Chem. Soc., 1979, 1 0 1 , 3847.
33. J. R. Fox, W. L. Gladfelter, G. L. Geoffroy, I. Tavanaiepour, S. Abdel-Mequid and V. W. Day, In o rg . Chem., 1981, 2 0 , 3230.
34. J. A. Iggo, M . J. Mays, P. R. Raithby and K. Hendrick, J. Chem. Soc., D a lto n Trans., 1983, 205.
35. C. Bianchini, A. M eli, M . Peruzzini, P. Frediani, M. A. Esteruelas and L. A. Oro, O rganom etallics, 1992, 1 1 , 138.
36. P. Barbaro, C. Bianchini, M. Peruzzini, A. Polo and F. Zanobini, In o rg . Chim. A cta, 1994, 2 2 0 , 5.
37. R. F. Heck; "Organotransition Metal Chemistry", Academic Press, New York, 1974.
38. G. W. Parshall; "Homogeneous Catalysis", Wiley-Interscience, New York, 1980. 39. J. P. Collman, L. S. Hegedus, J. R. Norton and R. G. Finke; "Principles and Applications o f Organotransition Metal Chemistry", University Science Books, M ill Valley, Calif. 1987.
40. C. A. Tolman, J. Am. Chem. Soc., 1970, 9 2 , 2953 and 2956. 41. D. S. Bohle and W. R. Roper, O rganom etallics, 1986, 5, 1607.
42. P. M. Treichel, W. K. Dean and W. M . Douglas, J. Organom et. Chem., 1972, 4 2 , 145.
43. H. Knoll, H. Hennig, B. Walther, H.-C. Bottcher and D. J. Stufkens, In o rg . Chim. A cta, 1 9 9 3 , 2 1 0 , 33.
44. D. S. Bohle, T. C. Jones, C. E. F. Rickard and W. R. Roper, O rganom etallics,
1 9 8 6 , 5, 1612.
45. S. M . Rocklage, R. R. Schrock, M . R. Churchill and H. Wasserman, O rg a n o m e ta llics, 1982, 1 , 1332.
46. A. J. Carty, Adv. Chem. Ser., 1982, 163.
47. R. P. Rosen, J. B. Hoke, R. R. W hittle, G. L. Geoffroy, J. P. Hutchinson and J. A. Zubieta, O rg an o m e ta llics, 1984, 3, 846.
48. J. Powell, M . R. Gregg and J. F. Sawyer, In o rg . Chem, 1989, 2 8 , 4451. 49. W. Malisch and P. Panster, Ang. Chem Engl. Ed., 1977, 1 6 , 408.
50. A. A . Cherkas, L. H. Randall, S. A. Maclaughlin, G. N. M ott, N. J. Taylor and A. J. Carty, O rg an o m e ta llics, 1988, 7 , 969.
51. W. Ehrl and H. Vahrenkamp, J. Organomet. Chem., 1973, 6 3 , 389. 52. L. Straudacher and H. Vahrenkamp, Chem B er., 1976, 1 0 9 , 218. 53. G. Hogarth, J. Organom et. Chem., 1991, 4 0 7 , 91.
54. A. M . A rif, T. A. Bright, D. U. Heaton, R. A. Jones and C. M . Nunn, P olyhe d ro n, 1990, 9 , 1573.
55. N. Lugan, G. Lavigne, J.-J. Bonnet, R. Reau, D. Neibecker and I. Tkatchenko, J. Am. Chem. Soc., 1988, 1 1 0 , 5369.
56. S.-G. Shyu and A. W ojcicki, O rganom etallics, 1984, 3 , 811.
57. Y.-F. Yu, J. Gallucci and A. W ojcicki, J. Am. Chem. Soc., 1983, 1 0 5 , 4826. 58. P. Leoni, M . Pasquali, M . Sommovigo, F. Laschi, P. Zanello, A. Albinati, F. Lianza, P. S. Pregosin and H. Ruegger, O rganom etallics, 1993, 1 2 , 1702.
59. S. Rosenberg and G. L. Geoffroy, O rganom etallics, 1985, 4 , 1184. 60. M . Cowie and S.K. Dwight, Inorg. Chem., 1979, 1 8 , 1209.
61. R. J. Puddephatt, Chem. Soc. Revs., 1983, 99.
62. A. L. du Preez, I. L. Marais, R. J. Haynes, A. Pidcock and M. Safari, J. Chem. Soc., D a lto n Trans., 1981, 1918.
63. H. Einspahr and J. Donohue, Inorg. Chem., 1974, 1 3 , 1839.
64. M . A. Pinto, P. J. Sadler, S. Neilde, M. R. Sanderson, A. Subbiah and R. Kuroda, J. Chem. Soc., Chem. Commun., 1980, 13.

![Figure 2.1 X-ray structure of [Fe2(C0)g(|i-C0)(|i-dppm)]](https://thumb-us.123doks.com/thumbv2/123dok_us/9092992.1444060/45.595.62.536.51.599/figure-x-ray-structure-of-fe-c-dppm.webp)
![Figure 2.2 Infra-red spectrum of [Fe2 (CO)^(p-CO)()i-H)(u-PCy2)((i-dppm)] in DCM](https://thumb-us.123doks.com/thumbv2/123dok_us/9092992.1444060/47.595.44.533.44.780/figure-infra-red-spectrum-fe-pcy-dppm-dcm.webp)
![Figure 2.3 NMR Hydride Resonance of [Fe2 (CO)XM-CO)(p-H)(n-PCy2)(p-dppm)]](https://thumb-us.123doks.com/thumbv2/123dok_us/9092992.1444060/48.595.58.541.47.728/figure-nmr-hydride-resonance-fe-co-pcy-dppm.webp)
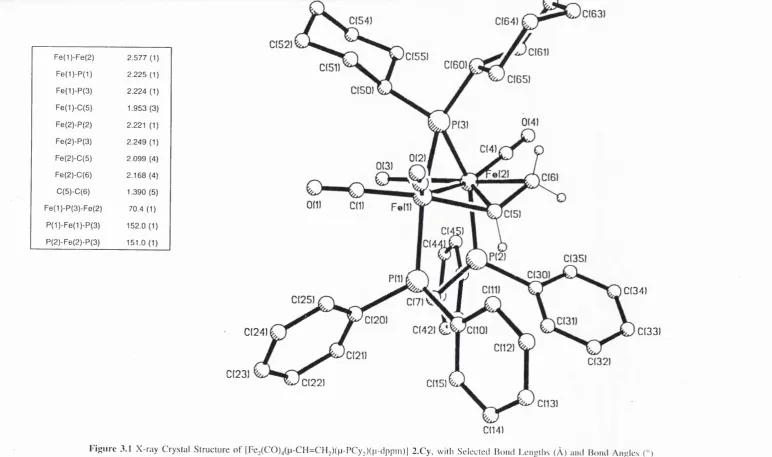
![Figure 2.5 X-ray Crystal Structure of [re2(C 0)/p-C 0)(p-H )(p-PC y2)(p-dppm )] l.C y , with Selected Bond Lengths (Â ) and Bond Angles (°)](https://thumb-us.123doks.com/thumbv2/123dok_us/9092992.1444060/52.843.309.699.41.471/figure-crystal-structure-selected-bond-lengths-bond-angles.webp)
![Figure 2.6 Schematic Diagram of [Fe2(CO)4(M.-CO)(fi-H)(p-PCy2)((i-dppm)]](https://thumb-us.123doks.com/thumbv2/123dok_us/9092992.1444060/53.595.203.338.267.399/figure-schematic-diagram-fe-co-co-pcy-dppm.webp)