Journal of Com DOI: 10.220
Abstract. β -problem in challenging structure pre to deal wit topology. He "BetaProbe1 β-sheet topo frequency o conformation interactions alignment pr pairwise inte the interacti pairwise alig programming programming probabilities the β-sheet t probability i observed con show that outperform method with Keywords: programming 1.Introducti
Proteins p organisms. B proteins is Therefore, it Further, the the structure Nuclear Mag costly, time addition, now primary stru tertiary struc determined b is a huge g structures an
Manuscript r 2016. Department o Ferdowsi Un The corresp eghdami.mah
mputer and Kno 067/cke.v1i2.
β-shee
Mah
sheet topology modern com intermediate diction. Diffe th the proble ere, ab-initio1" and "BetaP
ology. In thes of β-strand p n are spotte between β -robability, the eraction is con
ons. Furtherm gnment probab g approach is g optimizati of β-strand p topology. Mo is considered nformations f
BetaProbe1
the most rec respect to β-s
β-sheet t g; dynamic pr ion
perform criti Biologists be
determined t is important conventional of protein, n gnetic Resonan e-consuming,
w, from the uctures in the ctures of 30 by experimen gap between nd the number
received May 1
of Computer En niversity of Mas ponding author' [email protected]. owledge Engine 56005
et Topolo
hdie Eghda
y prediction i mputational
step toward erent methods em of determ
probability-ba
Probe2" are ut se methods, t pairwise inter ed. To pred -strand pairs e probability o nsidered to c more, to dete bility more ac s utilized. In
ion is com pairwise intera oreover, the β
to give bett for selection. E
and BetaP
cent integer sheet topology topology p rogramming; p
cal functions elieve that t by their t to specify th empirical m amely, X-ray nce (NMR) sp and sometim 30 million p e protein data 0 thousand o ntal methods [ the number r of determine
6, 2016; accept
ngineering, Eng shhad, Mashhad 's e-mail is: .ac.ir
eering, Vol. 1, N
ogy Pred
Intege
ami
, Tokta
s a major unre biology. It the protein t have been pr mining the ased methods tilized to spec the stability a raction and dict more fr
, besides p of occurring β
ompute the sc ermine the β
ccurately, a dy addition, the mbined with actions to det
β-sheet confor ter chances to
Experimental
Probe2 signif programming y prediction. prediction;
pairwise align
s within the he functiona
tertiary stru he protein str ethods to det crystallograp pectroscopy a mes impossib proteins with
abases [1], on of them have [2]. Therefore
of known p ed tertiary stru ted September 2
gineering Facul d, Iran. No. 1.
diction U
er Progr
am Dehgha
esolved t is a tertiary rovided β-sheet s called cify the and the β-sheet frequent airwise β-strand core of β-strand ynamic integer th the termine rmation o more results ficantly g-based integer nment; living ality of uctures. ructure. termine phy and are very ble. In known nly the e been e, there primary uctures. 24, lty, He com pro stru kno am β-s int dif ori ter two Th pai edg she wh clo the Fi mo stru she bon [4] cha exp val dim new [11Using P
ramming
ani, Mahm
ence, insuffici mputational oblem.One of the ucture is -s own as β-str mino acids lon strands and teraction betw fferent forms ( ientation given rmini [4]. Each o hydrogen b he interactions ired β-strands
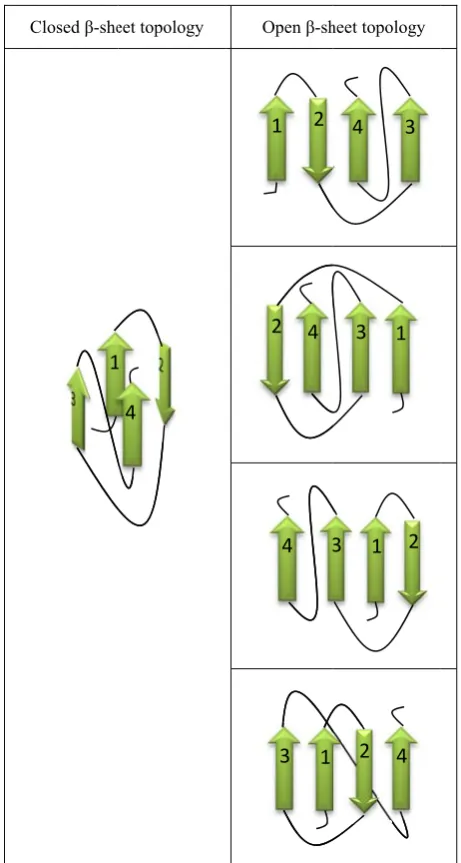
β-sheets can b ge strands an eets. Fig. 1 sh here four β-st osed ones a ci e first strand a
ig. 1. Open β-sh 1NZ0D. -st
β-sheet topol ost important ucture predict eet topology nd formations ]. Furthermor aracteristic o ploited [4].T luable infor mensional stru
w drugs [8], [ 1].
robabili
g
moud Naghi
ency of empir methods in
most freque sheet which
ands. -stran g [3] that inte make paired ween two β
(parallel or an n by the positi h amino acid i bonds with ot s between the
are known as be open or cl nd they are th
hows an exam trands interac
rcle is formed nd the last on
heet of a protein trands that form
sequenti
ogy predictio unresolved tion of protein
remains chal s between lin re, the global f β-sheet str he β-sheet t mation for ucture [6], [7]
[9] and determ
ity-base
ibzadeh
rical methods protein stru ent elements
consists of s nds are typica eract with ami
d β-strands
β-strands can nti-parallel) de
ion of the β-st in a β-strand c ther ones in e amino acid s a β-contact m losed. Open β
he most com mple of an op ct. On the ot d by a hydrog ne.
n with PDB (Pr m the β-sheet are
tial order.
on is regarde problems tow ns [5]. Correc llenging beca nearly distant l covariations ructures have topology pre predicting ], designing n mining foldin
ed
leads to utiliz ucture predict in the pro separate secti ally six to e ino acids of ot
(partners). n occur in
epending on th trands’ N- and can make at m the paired sta d residues of map.
β-sheets have mon types of pen β-sheet ty her hand, in gen bond betw
rotein Data Bank) e numbered in
ed as one of ward the tert
t prediction o ause of hydro
β-sheet resid s and constra e not been w
diction provi protein th new proteins ng pathways [
zing tion otein ions ight ther The two heir d C-most and. the two f β -ype,
the ween
) id
54 eghdami et.al: β-sheet Topology Prediction Using Probability-based Integer Programming
The main goal of predicting β-sheet topology from the protein’s amino acids is to determine the organization of β -strands in the β-sheets. This includes identifying β-strand members of each β-sheet and describing β-sheets by specifying paired β-strands and their interaction types. Further, β-contact maps are determined in β-sheet structure prediction. Different methods have been proposed to address the problem of predicting β-sheet topology which will be described in the next section.
In this article, we present BetaProbe1 [12] and
BetaProbe2, ab-initio probability based methods for β-sheet topology prediction. The main advantage of the proposed methods as compared to the previous researches is that we make use of the fact that more frequent and more stable conformations should have greater chances of being selected. For this purpose, the score of an interaction between each two β-strands is computed considering both pairwise alignment probability and pairwise interaction probability. Moreover, in order to make more accurate alignments, the β-strand optimum pairwise alignment is found using a dynamic programming approach. Furthermore, combining integer optimization with the β -strand pairwise interaction probability improves the accuracy of the predicted interactions. In addition, using β -sheet conformation probability in the last step of
BetaProbe1 leads to predicting more frequent and more stable conformations.
In the rest of this paper, first, related studies are reviewed in Section 2. Then, the details of the proposed methods will be described in Section 3. Finally, the performances of the proposed methods are compared with the most recent integer programming-based β-sheet prediction method in Section 4.
2. Related Work
Most β-sheet topology prediction methods utilize contact maps and strands alignment. Any improvement in the accuracy of these fields leads to a higher accuracy in determining the architecture of β-sheets. In this section first the related works in these fields are introduced. Then, some
β-sheet prediction methods are explained.
Specifying the protein contact map is the first step in determining its final structure. Mainly, a contact map is expressed by a two-dimensional matrix. For two amino acids ri and rj, if the value of the i-th row and the j-th column (0≤contact Map (i, j) ≤1) is closer to one then they are more likely to interact with each other in the final structure. In other words, the likelihood of their relationship in the final structure of proteins is higher. NNcon [13], DNcon [14], SVMcon [15] and Distill [16] can be mentioned as contact map prediction methods. CMAPpro [17], PSICOV[18] and PhyCMAP [19] are the most recent methods which include contact map prediction.
So far, methods with high accuracy and acceptable execution time have been suggested for the sequence alignment problem. Further, pairwise sequence alignment is the most common technique used in β-sheet prediction methods. The most usual approach to determine the best alignment between two strands is dynamic programming.
Many efforts have been made to address the problem of predicting β-sheet topology. These works can be divided into two major categories: homology-based methods and ab-initio methods. The homology-based methods such as SMURF [20], SMURFLite [21], and MRFy [22] use homological information of proteins for recognizing their topologies. On the other hand, ab-initio methods only consider amino acids’ pairing potentials and statistical information. In this article, we concentrate on the ab-initio
β-sheet topology prediction methods. They utilize different approaches such as statistical potentials[23], information theory[24], Bayesian models and exploration of entire search space[25], linear programming [5], [26], [27], hidden Markov models [28], and graph matching algorithms [4]. These approaches can be divided into two major categories[29]: in one category, all possible β -topologies are enumerated, and a score for each complete β -topology is computed. Then, the β-topology with the highest score is selected as the best one [7], [25]. In the other category, in order to predict the β-sheet topology of a protein, pseudo-energy is assigned to each pair of β-strands. Then the problem of determining the best β-topology is reduced to maximizing the strand-to-strand contact potentials of the protein [5], [4], [26], [27], [28], [30].
Journal of Computer and Knowledge Engineering, Vol. 1, No. 1 55
nature. On the other hand, some particular orientations are more favorable than others. In addition, models for computing the probability of open β-topologies for proteins were derived. The discriminative power of these models is reduced significantly because the number of possible β -strand organizations increase exponentially and there is not sufficient training data to reliably represent such conformations. Therefore, these models are limited to proteins that contain at most ten β-strands. In this research, we try to improve BCov by considering the stability and frequency of β-strand pairing and β-sheet conformation. 3. Proposed Method
In this article, two efforts are made to resolve the problem of predicting β-sheet topology: BetaProbe1 and
BetaProbe2.These efforts can predict both β-sheet topology and β-contact map. As previously mentioned, in BCov[26] two β-strands are paired based on only their alignment score; but, Ruczinski et al. [7] showed that the organization of β-strands into β-sheets is not random and there is a distinct pattern. Therefore, to improve BCov, we attempt to give greater chances to more stable and more frequent conformations during the selection. In this section, first, a general description of each attempt is presented. Then, the steps of the proposed methods are described in detail.
3-1. First Effort: BetaProbe1
BetaProbe1 consists of three major steps: (i) in order to achieve more accurate alignments, a dynamic programming approach is used to compute the β-strand pairwise alignment probability. In addition, pairwise interaction probability of each pair of β-strands is computed according to [32]. Then, both pairwise alignment probability and pairwise interaction probability are utilized to compute the score of each interaction (ii) to determine the maximum total strand-to-strand contact potentials of the protein an integer programming optimization is used. In this step, to enforce more stable and more observed paired β-strands to be selected, pairwise interaction scores obtained in the previous step are utilized (iii) the best β-sheet topology is achieved according to paired strands determined in the previous step. To predict more stable conformations, β -sheet topology probabilities are considered. The pseudo code of BetaProbe1 is illustrated in Pseudo code1.
Computing β-strand Pairwise Interaction Score: Many methods have been proposed to find the best alignment between sequences [33], [34]. Here we concentrate on an alignment method which is especially proposed for β -strands. In BetaProbe1 the alignment probability of each two β-strands is computed based on the proposed method in BetaZa[25]. In this method, the Needleman-Wunsch algorithm [33][34] is used to compute the optimum alignment between each pair of β-strands in the parallel and anti-parallel directions. Then, the probability of the optimum alignment is computed by dividing the score of the best alignment by the sum of all possible alignments. To improve the accuracy of the alignments, the amino acid
pairing potentials are used which are computed especially based on the β-amino acids.
Pseudocode 1: Probability-based algorithm for β-sheet topology prediction (BetaProbe1)
Input:protein’s strands
Output: an open β-sheet conformation with the highest probability
Step 1: Determining β-strand Pairwise Interaction Score for each pair of strands si and sjdo
compute their parallel and anti-parallel pairwise alignment probabilities
compute their parallel and anti-parallel pairwise interaction probabilities
scores=alignment probability × interaction probability
Step 2: Predicting the Closed β-Sheet Topology Solve the integer programming problem Step 3: Determining the Best Open β-Sheet Topology
for each closed β-topology do
for each interaction between two β-strands do Omit the interaction temporarily
Compute the probability of the new open β -sheet
Select the open β-sheet with the highest conformation probability.
To store the pairwise alignment probability, a matrix called "PAP (Pairwise Alignment Probability)" with n rows and 2n columns is defined. In this matrix, n is the number of β-strands in the protein. Matrix PAP is defined as follows:
PAP i,j =
Sparallel si,sj if i≤n and j≤n and j≠i
Santi-parallel si,sj if i≤n, n+1≤j≤2×n and j≠n+i
0 if j=i or j=n+i
(1)
In Equation (1), Sparallel (si,sj) represents the probability of optimum alignment between strands si, i=1,2,…,n, and sj, j=1,2,…,n, where their interaction type is parallel. Also, Santi-parallel (si,sj) represents the probability of optimum alignment between strands si, i=1,2,…,n, and sj, j=1,2,…,n, where their interaction type is anti-parallel. The definition shows that the matrix PAP is divided into two sections with an equal number of columns. The left section is used to store the parallel alignment probabilities and the right section is used to store the anti-parallel ones. The Score matrix for the protein in Fig. 1 is shown in Fig. 2-(a). It is important to note that the alignment probability depends on the spatial ordering of strands [25]. Therefore, the score of the optimum alignment between non-bridge strands can be different. This is expressed in (2) and (3):
56
Santiparallel (si,sj)
According and they ar compared to (Pairwise Int pairwise inte derived by [3 Matrix PIP c (4):
PIP i,j =
Pp
Pan
0
In (4), Ppar and sj to ma based on th status and th strands. Sim strands si an spatial order pairwise inte (6):
Pparallel(si,sj) =
Pantiparallel(si,sj)
In Fig. 2-( 1NZ0D. The of β-strands score, both alignment pr the interacti "Score" is int the protein. T Score i,j =PA In the m represent the last n colum note that the depends on illustrated in Prediction of in the integer probabilities paired β-stra model of B
BCov's. As a by solving th
maximize:
subject to:
(a)
)≠Santiparallel(sj,s
g to [32], som re more freq
others. Base teraction Prob eraction proba
32] were use contains n row
parallel si,sj ntiparallel si,sj
rallel(si,sj) repre ake a parallel he protein ch
he number of milarly, Pantipa
nd sj to make ring of strand eraction probab
Pparallel(sj,si)
) = Pantiparallel(sj,
(b), the matrix en the scores
are determin pairwise inte robability is c ions, a bi-d troduced, whe The matrix de AP(i,j)×PIP(i,j) matrix Score e scores of pa mns show anti
e score of an their spatial Fig.2-(c) for of the Closed β
r optimization are considere ands. In add
BetaProbe1 i a result, the cl he integer prob
Score(i
2×n
j=1 n
i=1
c1: X i,j ∈ 0,1 c2: X i,j +X j, ∀ c3: ∑2×nX(i,j)
j=1
c4: ∑n X i,j
i=1
c5: ∑2×nX i,j
j=1
c6: X i,i =X si)
me β-strand p quently obser d on this obse bability)" is d abilities of β -ed to compute ws and 2 n co
if i≤
if i≤n, n+1≤
esents the pro interaction in haracteristics f residues betw
arallel(si,sj) is e an antipara ds has no eff
bility. This is
si)
x PIP is comp of interaction ned. In the c eraction proba considered. To imensional n ere n is the nu finition is dec 1≤i≤n , 1≤j≤2× definition, th arallel interac i-parallel one n interaction b
ordering. T the protein 1N
β-Sheet Topo n problem the ed in order to dition, the in is defined d losed β-sheet blem in (8).
i,j) X(i,j)
1∀ 1≤i≤n , 1≤j ,i +X i,j+n +X ∀ 1≤i≤n, 1≤j≤2
∈ 0,1∀1 i n
+X i,j+n ∈{ +∑n (X j,i +
j=1
∀1 i n
i,i+n =0 ∀ 1≤i
eghdami et.
airs are more ved in natur ervation, matri defined to sto strands. The m e these probab olumns as def
≤n and j≤n and j≠ ≤j≤2×n and j≠i+ if j=i or j=i+n
obability of str n the final st such as the ween each tw the probabi llel interactio fect on the β
expressed in
puted for the ns between ea computation o
ability and p o store the sc n×2n matrix umber of β-str clared in (7).
×n
he first n co ctions. Similar
s. It is impor between two he Score ma NZ0D. ology: Unlike
pairwise inte o predict more nteger progra ifferently fro topology is ob
≤2×n
X j,i+n∈0,1 ×n
n
{0,1} ∀1 j n +X(j,i+n))∈ 1,2
≤n
al: β-sheet Top
(3) e stable
re, as rix "PIP ore the models bilities. fined in ≠i n n (4)
rands si tructure helical wo beta lity of on. The β-strand (5) and (5) (6) protein ach pair of each airwise cores of called rands in (7) olumns rly, the rtant to strands atrix is BCov, eraction e stable amming om the btained (8) 2 0 (b) (c) 0 F com ma inte 1N pro X zer stra bet con par wit c5) top (a) X = (b) Fi (b) pology Predictio 0 0 0 0 0.15 0.025 0.027 0 PIP = 0 0 0.01 0.27 0 0.27 0 0 0 0 0 0.15 0.025 0.027 0
Fig 2. (a) The mputed by the atrix PIP for pr
eraction probab NZ0D compute obability and pa
X is a n 2n b ro entries sh
ands. c2 co tween two str nstraints ensu rtner on eithe th at least one ).
In Fig. 3, the pology for pro
=
0 0 0 0 1 0 0 0
g. 3. (a) The m ) The predicted
parallel alignm
paral
parallel intera
paralle
on Using Proba
PA 0.138 0.019 0.023 0
0 0.238 0.437 0
0.01 0.27 0.2 0 0.01 0.2 0.01 0 0.2 0.27 0.27 0
Sco 0.138 0.019 0.023 0
0 0.238 0.437 0
matrix PAP fo dynamic progr rotein 1NZ0D bilities in [32]. ed by conside airwise interacti
binary matrix how an inter onstraint show
rands is paral ure that all str er side. Furth e and at most t matrix X and otein 1NZ0D a
0 0 0 0 0 0 0 0 0 1 0 0
atrix X obtained closed β-sheet ment score llel interaction action score el 1 4 ability-based Int AP= 0 1 1 0 0.05 0.015 0.015 0.055
27 0 0.99
27 0.99 0 27 0.73 0.99 0 0.73 0.7
ore=
0 1
1 0
0.05 0.015 0.015 0.055
or a protein wit ramming algor computed by (c) The matrix ering both p ion probability
x (constraint c raction betwe
ws whether llel or antipa rands have at hermore, each
two other β-st nd the predicte
are shown.
1 0 0 0 0 1 0 0 0 0 0 0
d by solving th topology for th anti-parallel a anti-par anti-par anti-parallel teger Programm 0.047 0.00 0.019 0.08
5 0 0
5 0 0
9 0.73 0.73 0.99 0.73
9 0 0.73
3 0.73 0
0.047 0.00 0.019 0.08
5 0 0
5 0 0
th PDB ID 1NZ rithm [25]. (b)
using the pairw x Score for pro airwise alignm in this paper.
c1) in which n een two rela the interact rallel. c3 and most one str h strand can p
trands (constr ed closed β-sh
0 1 0 0
Journal of Com
Determining previous step are determin predicted int each strand method for probabilities probability o the most pr topology. To the interactio process of d illustrated in open β-sheet
Fig. 4. The pro
mputer and Kno
g the Best O p, paired β-st ned by the eractions mak has two pa
the open determined b of each possib robable one o enumerate a ons of the clo determining th n Fig.4. In ad ts for the close
ocess of determ
owledge Engine
Open β-Sheet trands and th
integer prog ke closed β-sh artners. To ex
β-sheets, the by [32] are us ble open shee
is selected a all possible op osed one is om he best open ddition, Fig. 5 ed one in the F
mining the most
eering, Vol. 1, N
t Topology: eir interaction gram solution heets, in other
xtend the pr e β-sheet to sed. In this st t is computed as the best pen β-sheets, mitted at a tim
β-sheet topo 5 shows all p Fig. 3-(b).
probable open No. 1
In the n types n. The
words, roposed opology tep, the d. Then
β-sheet one of me. The logy is possible
β-sheet 3-2 Be Be
con pai alig com (ii) top int def In pai gre stra top clo Pse
Fig sho
2. Second Effo taProbe2 con
taProbe1, the nsidering bo irwise interac gnments, a d mpute the alig ) to unravel pology, an troduced. Unl
fined to maxi this step, bo irwise alignm eater chance t
ands for sel pology achiev osed. The pse eudocode 2.
Closed β-she
g. 5. All possibl ows the best ope
1
ort: BetaProbe
nsists of two e score of eac oth pairwise ction probabil dynamic progr gnment probab
the problem integer pro like BetaPro
imize the prod oth pairwise ment probabil
o more stable lection. Unlik ved by the int
eudo code of
eet topology
le open β-sheets en β-sheet topo
4
e2
o major step ch interaction alignment lity. To obtai ramming app ability of each m of determin
ogramming
obe1, the inte duct of the in interaction p lities are uti e and more ob
ke BetaProb
nteger program f BetaProbe2
Open β-sh
s from a closed ology for protein
1 2
4 3
2 4
3 1
s: (i) similar n is computed probability in more accu proach is used
pair of β-stra ning the β-sh
optimization eger problem nteraction sco probabilities ilized to giv bserved paired
be1, the β-sh m solution is is illustrated
heet topology
d one. The gray n 1NZ0D.
4 3
3 1 2
3 1
2 4
57
r to d by and urate d to ands
heet is m is
ores. and e a d β -heet
not d in
58
Pseudocode 2: prediction(Be
Input: pr Output:
probabi Step 1:
for
Step 2: for
So
Computing β
to BetaProbe
are compute interaction be Determining specifying th optimization interaction b strand intera several inter probabilities. maximize th scores, becau probability o strands accor the pairwis interaction s function is as the logarithm maximizing determining problem repr problem are final β-sheet
maximize:
i
subject to: c c c c c c : Probability-ba etaProbe2) rotein’s strands an open β-she ility
determining B-r each pair of st compute their pairwise compute their
pai scores=align pro Prediction of th r each pair of st
scoresne
olve the integer p
β-strand Pair e1, first the el ed as in Be
etween each p g the β-shee he best β-sheet . By assumin between two s actions, the p ractions is co . Therefore, a he product o use each pair of occurrence rding to the p se interaction scores have p
scending, it is ms of the pair their prod the β-sheet to resented in (9 the same as ( topology for p
logScor
2×n
j=1 n
i=1
c1: X i,j ∈ 0,1 c2: X i,j +X j,i
∀ c3: ∑2×nX(i,j)
j=1 ∈
c4: ∑n X i,j +
i=1
c5: ∑2×nj=1X i,j + c6: X i,i =X i,
ased algorithm f
s
eet conformatio
-strand pairwis trands si and sj
r parallel and a e alignment pro r parallel and a irwise interactio ment probabilit obability
he β-Sheet Topo trands si and sj
ew=log(scores)
programming p
rwise Interac lements of ma
etaProbe1. T pair of β-stran
et Topology: t topology is r ng that the e strands is inde probability of omputed by t an integer opt of β-strand p rwise interacti e of an inter
airwise alignm n probability positive value
s possible to m rwise interacti duct. Then, opology becom 9). Note that th 8). In Fig. 6, t protein 1NZ0
re(i,j) X(i,j)
∀ 1≤i≤n , 1≤j≤
i +X i,j+n +X ∀ 1≤i≤n, 1≤j≤2×
∈ 0,1∀1 i n
+X i,j+n ∈{0 +∑nj=1(X j,i +X
∀1 i n
i+n =0 ∀ 1≤i≤
eghdami et.
for β-sheet topo
on with the hig
e Interaction Sc do anti-parallel babilities antiparallel on probabilities ty×interaction ology do problem
ction Score: S atrices PIP an Then, the sc nds is determin
: The probl reduced to an event of exist ependent of o f the occurre the product o timization is u pairwise inte ion score sho action betwee ment probabil y. Since p s and the log maximize the ion scores ins the proble mes an integer he constraints the matrix X a
D are present
≤2×n
X j,i+n∈ 0,1 ×n
0,1} ∀1 j n X(j,i+n))∈ 1,2
≤n
al: β-sheet Top
ology ghest core s Similar nd PAP core of ned. lem of integer ting an other β -ence of of their used to eraction ows the en two lity and airwise garithm sum of stead of em of r linear s of the and the ted. (9) (a) (b) Fig pro 4. R In are and Ev pro (12 sta Pre Rec F1-N po Da Th 91 fol ass inc DS out Cr fol are set is r me acc W a 1 Be stra sim wit pology Predictio X = 0 0 0 0
g 6. (a) matri ogramming prob
Results this section, f e described. T d BetaProbe2 valuation met oposed metho 2) are used. T ate-of-the-art m ecision= Tp+FPTP
call= TP+FNTP ×1 -score= 2×PrecisPrecisi Note that TP, sitives, and fa ataset:We us his dataset is e
6 proteins. To lds randomly signing the se cludes: (1) th SSP) and (2) t
tput).
ross validation ld is consider e the training s t. Predictions
repeated for a easures are complished.
We carried ou 10-fold cross-v etaSheet916 fo
ands and less mulation, the p
th less than or 3
on Using Proba
0 0
0 0
0 0
0 1
ix X represent blem. (b) the fin
first, the evalu Then, the resu are presented trics: To eva ods, well-know
These metrics methods [5], [2
×100
00
sion×Recall on+Recall
, FP, and FN alse negatives
ed the BetaSh extracted from o perform cros
and evenly. D econdary struc he extended β
he isolated β-b
n: At each st ed as the test set. Models ar are determine all proteins in computed ut three simul validation exp or proteins wit than three par proposed meth r equal to five
4
3
parallel
ability-based Int
0 0 1
0 0 0
0 0 0
0 0 0
nt the result o nal predicted β
-uation metrics ults of evalua d.
aluate the per wn metrics i s have been [26], [25]: represent tru values, respec heet916 set fo m the PDB by ss-validation, DSSP program
cture. In this
β-strands (sho bridges (show
tep in a cros t data and the re trained base ed in the test
the original s after the lations. In the periment was ith less than o rtners. Simila hod was evalu e β-strands wi
1 anti-pa teger Programm 0 0 0 0 0 0 0 0
of solving int -sheet topology
s and the data ating BetaPro
rformance of n (10), (11)
used to evalu
ue positives, f ctively. or the evaluat
y [4]. It inclu it is split into m [35] is used
article β-resid own by E in wn B in the DS
ss-validation, e remaining o ed on the train set. This proc set. The accur predictions e first simulat
performed on r equal to fou rly, in the sec uated on prote ith less than th
2 arallel ming eger y a set obe1 the and uate (10) (11) (12) false tion. udes 10-d for dues the SSP one ones ning cess racy are tion, n the ur β -cond
Journal of Com
partners. The with less tha partners. BetaProbe state-of-the-a integer progr BCov, the BetaPro wer the same data
In Table 1 level is com recall, precis table.
Wilcoxon determine w precision, re perform the subsidiaries the results o these subsets 5%, there is the two meth with up to s recall improv BCov is sign concluded probabilities, in the comp combining improves the Chart 2, and
BetaProbe1
compared to Table 1. The proteins with 6
Evaluation le
strand pairin
Pairing direct
a)The evaluati b) The evaluat c) The evaluat
mputer and Kno
e third simul an or equal to
e1 and BetaP
art method, B ramming. For
residue pair re used. Then
a set. 1, the perform mpared with sion, and F1-s test for relat whether there ecall, and F1 e test, the d
as declared in of BetaProbe
s. The test sho a significant hods at the pa
ix and up to vement of Be
nificantly mea that besides , considering mputation of it with the e accuracy of d Chart 3 the at pairing d BCov's. e performance
6 or fewer β-str
evel Metho
ng BCov BetaProbe BCov BetaProbe BCov BetaProbe tion BCov BetaProbe BCov BetaProbe BCov BetaProbe
ion is done on p tion is done on tion is done on p
owledge Engine
ation was per six β-strands
Probe2 were BCov, which r this purpose ring probabil n the methods mance of BetaP
the performa score measure ted samples h is a significa -score of the data set wa n the Dataset
1 and BCov owed that with t difference b
airing directio five strands.
etaProbe1 com aningful. From s using β-s
pairwise inte
β-strands int integer pr f pairing dire recall, precisi direction leve of BetaProbe rands on BetaSh
od Recall
6 a 79
e1 6 73
5 b 81
e1 5 76
4 c 82
e1 4 75
6 64
e1 6 70
5 64
e1 5 73
4 72
e1 4 74
proteins with up proteins with u proteins with u
eering, Vol. 1, N
rformed on p with less tha compared w h is also bas e, in the first
lities calcula s were evalua
Probe1 at the ance of BCo es are shown has been util ant difference e two metho s broken in t section. Afte
were evalua h an average e
etween the re on level for p This means t mpared with m Table 1, it sheet confor eraction proba teraction scor
ogramming ections. In C
ion, and F1-sc el is illustrate e1 at strand le heet916.
Precision F1
84 69 86 73 85 72 68 67 69 70 75 70
p to 6 β-strands up to 5 β-strands p to 4 β-strands
No. 1 proteins an three with the sed on step of ated in ated on e strand ov. The in this ized to e in the ods. To nto ten er that, ated for error of ecall of proteins that the that of can be rmation abilities re and greatly Chart 1, core of ed and evel on -score 82 71 83 74 83 74 66 68 66 72 73 72 s s to the me Fu stra sho sig Be pro im com
In Table 2, th BCov at stran e precision an ethod at pair urther, the prec
and pairing owed that w gnificant diffe
taProbe2 at t oteins. It c mprovement of mpared with B
Chart 1: R
Chart 2: Pre
Chart 3: F1 BCov BetaProbe BCov BetaProb BCov BetaProb he performanc nd level. For nd the F1-sco ing direction cision of Beta
level. Wilcox with an avera
erence betwee the pairing dir can be con f BetaProbe2
BCov's. Recall compariso ecision compari -score comparis 58 60 62 64 66 68 70 72 74 76 Recall(%) 1 62 64 66 68 70 72 74 76 Precision(%) e1 6 6 6 6 7 7 7 F1-score(%) be1
ce of BetaPro
the three sub ore measures n level is be
aProbe2 is bet xon test for age error of en the precisi
rection level ncluded that
is significant
on of BetaProb
ison of BetaPro
son of BetaPro
64 64
70
up to 6 up
Numbe 6867 2 4 6 8 0 2 4 6
up to 6 u
Numbe 6668 62 64 66 68 70 72 74
up to 6
Numb
obe2 is compa bsets of prote of the propo etter than BC tter than BCo
related samp 5%, there i on of BCov for all subset the precis tly meaningfu
be1 to BCov
obe1 to BCov
obe1 to BCov 4
72
73 74
to 5 up to 4
er of strands
69
75
70 70
up to 5 up to
er of Strands
66 73
72 72
up to 5 up to
er of Strands
60
The reason compared w probabilities enforces β-st the nature t addition, to strands, a dy which gaps a pairing poten has improved Table 2. T
proteins
Evaluation le
strand pairin
Pairing direct
In Table3 compared to respectively. methods. Bet best β-sheet
BetaProbe2 BetaProbe2
the precision contact map the recall, pr at pairing dir are represen respectively. In Table5 and BetaPro
strand level
BetaProbe1, in the intege product of th predicting
BetaProbe2 b range of zero interactions precision of p decreases.
n for the impr with BCov is to the integer trand interact to be selecte improve the ynamic progr are allowed. F ntials provide d the accuracy The performance s with 6 or fewe
evel Metho
ng
BCov
BetaProbe
BCov
BetaProbe
BCov
BetaProbe
tion
BCov
BetaProbe
BCov
BetaProbe
BCov
BetaProbe
3 and Table4 o BetaZa at th The same ali taZa searches t topology. A
is less th is better at pa n of BetaPro
level is comp recision, and F
rection level w nted in Ch
and Table6
obe2 are repr l, respective the sum of th er programmin he interaction
fewer intera because the sc o and one. Sub are the mos predicted inte
rovement of p that adding r programmin tions which a ed with high pairwise alig ramming app Furthermore, u ed by the Bet y.
e of BetaProbe er β-strands on
od Recall
6 79
e2 6 65
5 81
e2 5 68
4 82
e2 4 71
6 64
e2 6 63
5 64
e2 5 67
4 72
e2 4 71
4 the results he residue lev ignment techn the entire sea Although the han BetaZa,
airing directio
obe2 at stran parable with B
F1-score mea with the other
art4, Chart the performa resented at th ely. As men he interaction
ng step while n scores is ma actions betw cores of the in bsequently, th st frequent o eractions incre
eghdami et.
proposed meth pairwise inte g in the secon are more frequ her probabilit
gnments betw proach is utili using the amin
aZa in the fir
2 at strand leve Beta Sheet 916
Precision F1
84
85
86
87
85
90
68
83
69
87
75
89
of BetaProb
vel and strand nique is used arch space to f execution ti the precisi on level. In ad
d pairing lev BetaZa's. Com
sures of BetaP
r method, the 5, and Ch ance of BetaP
he residue lev ntioned befo scores is max e in BetaProb
aximized. It le ween β-stran
nteractions are he predicted p ones. Therefo eases while the
al: β-sheet Top
hods as eraction nd step, uent in ties. In ween β
-ized in no acid rst step
el on 6.
-score
82
73
83
76
83
79
66
72
66
75
73
79
be2 are d level,
in both find the ime of ion of ddition, vel and mparing
Probe2
results hart 6,
Probe1
vel and ore, in ximized
be2 the eads to nds in
e in the airwise ore the
e recall E
E
P
pology Predictio
Table 3. The p proteins wi
Evaluation level
Contact map
Table 4. The p proteins wi
Evaluation level
strand pairing
airing direction
Chart 4: Recal BCov
BetaZa BetaProb
on Using Proba
performance of ith 6 or fewer β
Method
BetaZa 6
BetaProbe2 6
BetaZa 5
BetaProbe2 5
BetaZa 4
BetaProbe2 4
performance of th 6 or fewer β
-Method
BetaZa 6
BetaProbe2 6
BetaZa 5
BetaProbe2 5
BetaZa 4
BetaProbe2 4
BetaZa 6
BetaProbe2 6
BetaZa 5
BetaProbe2 5
BetaZa 4
BetaProbe2 4
l comparison of 0 20 40 60 80 100
Recall(%)
e2
ability-based Int
f BetaProbe2 at
β-strands on Bet
Recall Pre
78
6 58
80
5 60
82
4 61
fBetaProbe2 at -strands on Bet
Recall Pr
83
6 65
87
5 68
91
4 71
80
6 63
84
5 67
88
4 71
f BetaProbe2 to up to 6 up
Number
teger Programm
strand level on ta Sheet 916
ecision F1-sco
80 79
80 67
80 80
79 68
82 82
80 69
strand level on ta Sheet 916.
ecision F1-sco
84 84
85 73
88 87
87 76
91 91
90 79
81 81
83 72
85 84
87 75
88 88
89 79
o other method to 5 up to 4
of Strands
ming
n
re
n
ore
Journal of Com
Chart 5: Prec
Chart 6: Prec
5. Conclusio The issue considered
BetaProbe1
methods for t In these me probabilities programming allowed. Th interaction i interaction is probability a reduced the integer optim are perform results show integer pro novelties in t 1. Consideri pairwise an interac 2. Combinin interactio 3. Consideri nature to 4. Consideri in the inte 5. The abili
sheet stru 6. The abili sheet topo BCov BetaZ BetaP BCov BetaZ BetaP
mputer and Kno
cision comparis
cision comparis
on and Future e of determin
as a challen and BetaP
the β-sheet to thods, first, t
of β-strands a g approach w hen, the prob
is computed. s computed u and pairwise in problem of fi mization. 10-f med to evalua that these me ogramming-ba this research c ing both pair interaction pr ction between ng the prob on with the int
ing β-sheet predict more ing the spatia eger programm ity of the pro ucture for prot ity of the pro ology for prot 1 Precision(%) Za Probe2 1 F1-score(%) Za Probe2 owledge Engine
son of BetaPro
son of BetaPro
e Work ning the topo
nging proble
Probe2, two opology predic the optimum are determine while any nu ability of th . After that, utilizing both
nteraction pro inding the β-s fold cross-val ate the propo ethods outperf ased method can be summa rwise alignm robability to c n two β-strands
bability of teger program
conformation frequent β-top l ordering of ming; oposed metho
teins with mul oposed metho teins with clos
0 20 40 60 80 00
up to 6
Numb 0 20 40 60 80 00
up to 6
Numb
eering, Vol. 1, N
be2 to other me
be2 to other me
ology of β-sh em. In this o probability
ction, are intro pairwise alig ed using the dy umber of ga e occurrence , the score pairwise alig obability. Fina
sheet topology lidation exper osed method form the most d[26]. The arized as follow ment probabili
compute the s s;
occurrence mming;
probability pologies;
β-strands in β
ds to predict ltiple β-sheets
ds to predict sed β-sheets.
up to 5 up to
ber of Strands
up to 5 up to
ber of Strands
No. 1 ethods ethods heets is paper, y-based oduced. gnment ynamic aps are of an of on gnment ally, we y to an riments ds. The t recent major w: ity and score of of an in the β-sheets t the β -s; t the β
-fur PS Ou stra to hig new E T E P Re [1] [2] [3] o 4 s o 4 s The performa rther. By com SICOV [18] on
ur methods c ands with less
predict protei gher order par w constraints Table 5. The p residue level o
Evaluation level
Contact map
Table 6. The pe strand level o
Evaluation level
strand pairing
Pairing direction
eferences ] J. Peng,
protein Toyota T ] C. W. O
sheet pr of Techn ] M. J. S
conform
ance of predi mbining resid nes, the metho an predict pr s than three p ins with a hig rtners by exten to the integer erformance of B on proteins with Shee Method BetaProbe1 6 BetaProbe2 6 BetaProbe1 5 BetaProbe2 5 BetaProbe1 4 BetaProbe2 4
erformance of B on proteins with Shee Method BetaProbe1 6 BetaProbe2 6 BetaProbe1 5 BetaProbe2 5 BetaProbe1 4 BetaProbe2 4 BetaProbe1 6 BetaProbe2 6 BetaProbe1 5 BetaProbe2 5 BetaProbe1 4 BetaProbe2 4 , “Statistical structure p Technological O’Donnell, “ oteins,” PhD nology, 2011. Sternberg and mation of pro
ictions can be due pairing p ods can becom roteins with partners. This gher number nding probabi r programming BetaProbe1 and h 6 or fewer β-s et 916.
Recall Pre
6 63
6 58
5 64
5 60
4 64
4 61
BetaProbe1 and h 6 or fewer β-s et 916.
Recall Pre
6 73
6 65
5 76
5 68
4 75
4 71
6 70
6 63
5 73
5 67
4 74
4 71
inference fo prediction,” D
l Institute at C “Ensemble m
thesis, Massa d J. M. Tho oteins: an an
e improved e propensities w me more accur
six or fewer s can be exten of β-strands ilities and add g step. d BetaProbe2 a strands on Beta
ecision F1-scor
66 64
80 67
66 65
79 68
67 65
80 69
d BetaProbe 2 a trands on Beta
ecision F1-scor
69 71
85 73
73 74
87 76
72 74
90 79
67 68
83 72
70 72
87 75
70 72
89 79
r template-ba Doctoral the Chicago, 2013 odeling of b achusetts Insti ornton, “On nalysis of b
61
beta-62 eghdami et.al: β-sheet Topology Prediction Using Probability-based Integer Programming
pleated sheets,” J. Mol. Biol., vol. 110, no. 2, pp. 285–296, 1977.
[4] J. Cheng and P. Baldi, “Three-stage prediction of protein β-sheets by neural networks, alignments and graph algorithms,” Bioinformatics, vol. 21, no. suppl 1, pp. i75–i84, 2005.
[5] A. Subramani and C. A. Floudas, “Beta-Sheet Topology Prediction With High Precision and Recall for Beta and Mixed alpha/beta Proteins,”
PLoS One, vol. 7, no. 3, 2012.
[6] S. M. Zaremba and L. M. Gregoret, “Context-dependence of Amino Acid Residue Pairing in Antiparallel β-Sheets,” J. Mol. Biol., vol. 291, no. 2, pp. 463–479, 1999.
[7] I. Ruczinski, C. Kooperberg, R. Bonneau, and D. Baker, “Distributions of beta sheets in proteins with application to structure prediction,” Proteins Struct. Funct. Bioinforma., vol. 48, no. 1, pp. 85–97, 2002. [8] T. Kortemme, “Design of a 20-Amino Acid, Three-Stranded -Sheet Protein,” Science, vol. 281, no. 5374, pp. 253–256, Jul. 1998.
[9] B. Kuhlman, G. Dantas, G. C. Ireton, G. Varani, B. L. Stoddard, and D. Baker, “Design of a novel globular protein fold with atomic-level accuracy,”
Science, vol. 302, no. 5649, pp. 1364–1368, 2003. [10] J. S. Merkel and L. Regan, “Modulating protein
folding rates in vivo and in vitro by side-chain interactions between the parallel β strands of green fluorescent protein,” J. Biol. Chem., vol. 275, no. 38, pp. 29200–29206, 2000.
[11] Y. Mandel-Gutfreund, S. M. Zaremba, and L. M. Gregoret, “Contributions of residue pairing to β -sheet formation: conservation and covariation of amino acid residue pairs on antiparallel β-strands,”
J. Mol. Biol., vol. 305, no. 5, pp. 1145–1159, 2001. [12] M. Eghdami, T. Dehghani, and M. Naghibzadeh,
“BetaProbe: A probability based method for predicting beta sheet topology using integer programming,” in Computer and Knowledge Engineering (ICCKE), 2015 5th International Conference on, 2015, pp. 152–157.
[13] A. N. Tegge, Z. Wang, J. Eickholt, and J. Cheng, “NNcon: improved protein contact map prediction using 2D-recursive neural networks,” Nucleic Acids Res., vol. 37, no. suppl 2, pp. W515–W518, 2009. [14] J. Eickholt and J. Cheng, “Predicting protein
residue–residue contacts using deep networks and boosting,” Bioinformatics, vol. 28, no. 23, pp. 3066–3072, 2012.
[15] J. Cheng and P. Baldi, “Improved residue contact prediction using support vector machines and a large feature set,” BMC Bioinformatics, vol. 8, no. 1, pp. 113–121, 2007.
[16] D. Baú, A. J. M. Martin, C. Mooney, A. Vullo, I. Walsh, and G. Pollastri, “Distill: a suite of web
servers for the prediction of one-, two-and three-dimensional structural features of proteins,” BMC Bioinformatics, vol. 7, no. 1, p. 402, 2006.
[17] P. Di Lena, K. Nagata, and P. Baldi, “Deep architectures for protein contact map prediction,”
Bioinformatics, vol. 28, no. 19, pp. 2449–2457, 2012.
[18] D. T. Jones, D. W. A. Buchan, D. Cozzetto, and M. Pontil, “PSICOV: precise structural contact prediction using sparse inverse covariance estimation on large multiple sequence alignments,”
Bioinformatics, vol. 28, no. 2, pp. 184–190, 2012. [19] Z. Wang and J. Xu, “Predicting protein contact map
using evolutionary and physical constraints by integer programming,” Bioinformatics, vol. 29, no. 13, pp. i266–i273, 2013.
[20] A. Kumar and L. Cowen, “Recognition of beta-structural motifs using hidden Markov models trained with simulated evolution,” Bioinformatics, vol. 26, no. 12, pp. i287–i293, 2010.
[21] N. M. Daniels, R. Hosur, B. Berger, and L. J. Cowen, “SMURFLite: combining simplified Markov random fields with simulated evolution improves remote homology detection for beta-structural proteins into the twilight zone,”
Bioinformatics, vol. 28, no. 9, pp. 1216–1222, 2012.
[22] N. M. Daniels, A. Gallant, N. Ramsey, and L. J. Cowen, “MRFy: remote homology detection for beta-structural proteins using Markov random fields and stochastic search,” Comput. Biol. Bioinformatics, IEEE/ACM Trans., vol. 12, no. 1, pp. 4–16, 2015.
[23] T. J. P. Hubbard, “Use of beta-strand interaction pseudo-potentials in protein structure prediction and modelling,” in System Sciences, 1994. Proceedings of the Twenty-Seventh Hawaii International Conference on, 1994, vol. 5, pp. 336– 344.
[24] R. E. Steward and J. M. Thornton, “Prediction of strand pairing in antiparallel and parallel beta sheets using information theory,” Proteins Struct. Funct. Bioinforma., vol. 48, no. 2, pp. 178–191, 2002.
[25] Z. Aydin, Y. Altunbasak, and H. Erdogan, “Bayesian models and algorithms for protein β -sheet prediction,” IEEE/ACM Trans. Comput. Biol. Bioinforma., vol. 8, no. 2, pp. 395–409, 2011. [26] C. Savojardo, P. Fariselli, P. L. Martelli, and R.
Casadio, “BCov: a method for predicting β-sheet topology using sparse inverse covariance estimation and integer programming,” Bioinformatics, pp. 3151–3157, 2013.
Journal of Computer and Knowledge Engineering, Vol. 1, No. 1 63
Comput. Biol. Bioinforma., vol. 5, no. 4, pp. 484– 491, 2008.
[28] M. Lippi and P. Frasconi, “Prediction of protein β -residue contacts by Markov logic networks with grounding-specific weights,” Bioinformatics, vol. 25, no. 18, pp. 2326–2333, 2009.
[29] R. Fonseca, G. Helles, and P. Winter, “Ranking Beta Sheet Topologies with Applications to Protein Structure Prediction,” J. Math. Model. Algorithms, vol. 10, no. 4, pp. 357–369, 2011.
[30] R. Rajgaria, Y. Wei, and C. A. Floudas, “Contact prediction for beta and alpha beta proteins using integer linear optimization and its impact on the first principles 3D structure prediction method ASTRO-FOLD,” Proteins Struct. Funct. Bioinforma., vol. 78, no. 8, pp. 1825–1846, 2010. [31] D. T. Jones, “Protein secondary structure prediction
based on position-specific scoring matrices,” J. Mol. Biol., vol. 292, no. 2, pp. 195–202, 1999. [32] I. Ruczinski, “Logic regression and statistical issues
related to the protein folding problem,” PhD thesis, University of Washington, 2000.
[33] S. B. Needleman and C. D. Wunsch, “A general method applicable to the search for similarities in the amino acid sequence of two proteins,” J. Mol. Biol., vol. 48, no. 3, pp. 443–453, 1970.
[34] O. Gotoh, “An improved algorithm for matching biological sequences,” J. Mol. Biol., vol. 162, no. 3, pp. 705–708, 1982.
[35] W. Kabsch and C. Sander, “Dictionary of protein secondary structure: pattern recognition of hydrogen bonded and geometrical features,”