The Relationship Between DNA Damage Response and Accumulation of DNA Damage in LRRK2 G2019S Parkinson’s Disease
Divya Mahesh
Honors Thesis
Department of Psychology and Neuroscience University of North Carolina at Chapel Hill
Dr. Laurie Sanders, Thesis Advisor Dr. Sabrina Robertson, Thesis Advisor Dr. Kathryn Reissner, Reader
Acknowledgements
Firstly, I would like to thank Dr. Laurie Sanders for providing me with the opportunity to work in her lab and for being a great mentor over the past three years. I have truly enjoyed learning more about neuroscience, Parkinson’s disease, and more throughout my time in the lab. I am also grateful to the members of the Sanders Lab, specifically Dr. Claudia Gonzalez-Hunt and Catherine Toste, for supporting me through every experiment, success, and failure. I largely credit my development as a scientist to the members of this lab.
Additionally, I would like to thank Dr. Sabrina Robertson for guiding me through the process of scientific writing and supporting me as I completed my thesis. I have learned so much from her through this whole process. I would also like to thank Dr. Kathryn Reissner for taking the time to serve on my Thesis Committee and participate in my defense.
I would like to thank Dr. Beth Kurtz-Costes and Dr. Keely Muscatell for providing insight into the process of conducting research and writing a thesis, as well as for taking the time to answer any questions that I have had. I am grateful for their weekly lectures, which facilitated this entire process.
Finally, I would like to thank my family and friends for believing in me and pushing me to achieve my goals. I am so grateful for their constant love, support, and encouragement throughout the past four years. I would not be the person I am today without them.
Abstract
Parkinson’s disease (PD) is a neurodegenerative disease that affects 7-10 million people worldwide. Among people with PD, an accumulation of DNA damage has been noted, with one possible explanation for this accumulation being a dysregulated DNA damage response. Studying this DNA damage can open doors for new therapeutic targets of PD. The goal of the current study was to evaluate the role of ataxia telangiectasia mutated (ATM), a protein involved in DNA damage response, and leucine rich repeat kinase 2 (LRRK2), a kinase protein, in the accumulation of DNA damage in PD. I hypothesized that the implication of ATM and LRRK2 activation in LRRK2 G2019S PD indicates a relationship between a DNA damage response through ATM, LRRK2 kinase activity, and the accumulation of DNA damage. Western blot and immunocytochemistry (ICC) experimental methods were used to test this hypothesis in healthy control and LRRK2 G2019S lymphoblastoid cell lines (LCLs). The results revealed increases in the phosphorylation of H2AX, a proxy for DNA double-strand breaks (DSBs), in LRRK2 G2019S cells. Increased baseline phosphorylation of ATM in LRRK2 G2019S cells as well as a decrease in H2AX phosphorylation following inhibition of ATM kinase activity suggested a relationship between a DNA damage response through ATM and the accumulation of DNA damage. Interestingly, the results also showed a reversal in DNA damage following inhibition of LRRK2 kinase activity, implicating LRRK2 kinase activity in the accumulation of DNA damage in PD. Ultimately, the results of this study indicate a relationship between the accumulation of DSBs in PD, the DNA damage response via ATM, and LRRK2 kinase activity, although the exact nature of this relationship is still unknown.
The Relationship Between DNA Damage Response and Accumulation of DNA Damage in LRRK2 G2019S Parkinson’s Disease
Introduction
Parkinson’s disease (PD) is a common neurodegenerative disease that affects 7-10 million people globally (Beitz, 2014). PD is characterized mainly by neurodegeneration of the nigrostriatal pathway. PD is associated with symptoms such as bradykinesia, rigidity, tremors, and cognitive impairment. Approximately 10% of PD cases can be attributed to genetics or family history, but most PD cases are of unknown etiology, though this can vary based on demographics (D. G. Hernandez, Reed, & Singleton, 2016).
Leucine-Rich Repeat Kinase 2 (LRRK2)
The LRRK2 gene codes for the large leucine-rich repeat kinase 2 (LRRK2) protein, which is also known as dardarin (Li, Tan, & Yu, 2014). LRRK2 is believed to be involved in cellular functions such as protein-protein interactions, vesicle trafficking and neurite growth, among others. It also has two primary enzymatic functions: the hydrolysis of GTP to GDP by the ROC-GTPase domain and the catalysis of phosphorylation by the protein kinase domain. The latter of these functions is of particular importance in PD, as is described below (Hardy, Cai, Cookson, Gwinn-Hardy, & Singleton, 2006).
One cause of both sporadic and familial PD are mutations of the LRRK2 gene (D. Hernandez et al., 2005). The most common mutation is the G2019S mutation, in which serine takes the place of glycine at the 2019 amino acid position of the LRRK2 protein. G2019S is associated with 1-7% of genetic PD cases and 1-3% of sporadic PD cases, although this varies based on demographics (Li et al., 2014). This mutation is located within the protein kinase domain of LRRK2, specifically within a region that may alter the catalytic role of LRRK2 (West et al., 2005).
Prior research has found that the G2019S mutation results in an increase in kinase activity, which could indicate a mechanism for the involvement of LRRK2 in PD (Luzon-Toro, Rubio de la Torre, Delgado, Perez-Tur, & Hilfiker, 2007).
Ataxia Telangiectasia Mutated (ATM)
Ataxia telangiectasia mutated (ATM) is a protein that is mostly located within the nucleus of cells, although it can exist in the cytoplasm as well (Shiloh, 2003). Along with its role in cell development and growth, ATM is involved in the cellular response to DNA damage (Shiloh, 2003). Similar to LRRK2, ATM is a protein kinase, with its primary catalytic function being the phosphorylation of various substrates following DNA damage. Substrates for ATM include proteins involved in DNA repair, such as P53 and CHK2 (Bradbury & Jackson, 2003). A schematic of the DNA damage response pathway involving ATM, CHK2, and P53 is shown in Figure 1 (Sulli et al., 2012). Mutations that affect the functionality of ATM can result in ataxia telangiectasia, a genetic disorder that impairs DNA repair pathways and results in motor dysfunctions (Shiloh, 2003). ATM mutations can also increase the risk of certain cancers (Khanna, 2000).
Although there is little research that directly connects ATM to LRRK2 G2019S PD, studies have revealed a link between ATM and models of PD involving alpha-synuclein, a protein that irregularly aggregates in some forms of PD. Past research has observed an increase in alpha-synuclein pathology in mice lacking ATM (Eilam, Peter, Groner, & Segal, 2003). Additionally, one study noted an increase in phosphorylated ATM in synucleinopathy models of PD, indicating alterations in the DNA damage response pathway in PD (Milanese et al., 2018). Due to its implication in DNA damage response in other models of PD, ATM is of interest in this study. Additionally, ATM is of interest due to prior research that indicates a relationship between ATM and LRRK2 (Chen et al., 2017). Past research suggests that LRRK2 phosphorylation depends on
ATM activity, but ATM phosphorylation does not appear to depend on LRRK2 activity (Chen et al., 2017). This study is of importance because it proposed a connection between ATM and LRRK2.
Nuclear DNA Damage
DNA damage occurs naturally in all cells as a result of stress, spontaneous processes, and exogenous sources, such as UV light or chemicals (Roos & Kaina, 2006). Cells respond to damaged DNA either by signaling DNA repair pathways or by initiating programmed cell death, also known as apoptosis. Without an appropriate response to DNA damage, severe consequences can ensue, including mutations, unregulated cell death, and diseases such as cancer.
There are several types of DNA damage, many of which are of interest in PD research. Single strand breaks (SSBs) entail damage to only one strand of DNA (Berquist & Wilson, 2012). SSBs are commonly repaired through a process called base excision repair (BER). Double strand breaks (DSBs) involve damage to both strands of DNA (McKinnon, 2013). DSBs are repaired via homologous recombination or non-homologous end joining (McKinnon, 2013). Homologous recombination uses the sister chromatid as a template to repair the break. Non-homologous end joining directly connects the broken sides of the damaged DNA strands via ligation. Although homologous recombination is more accurate, neurons do not undergo DNA replication as other cells do, making the presence of sister chromatids rare. This makes non-homologous end joining a more likely repair mechanism in neurons, even though it is more prone to error (McKinnon, 2013). However, a recent study suggests that homologous recombination can occur in post-mitotic neurons if an RNA template is present (Welty et al., 2018).
Soon after a DSB occurs, DNA damage response pathways involving ATM are activated. ATM is autophosphorylated at serine 1981 and it in turn phosphorylates its targets, such as P53
and CHK2 (Paull, 2015). This pathway is illustrated in Figure 1 (Sulli et al., 2012). Additionally, following activation of this DNA damage response pathway, H2AX, a histone protein and substrate of ATM, is phosphorylated at position Ser139 (Podhorecka, Skladanowski, & Bozko, 2010). Therefore, H2AX functions as an experimental proxy for DSBs. An impairment in this DNA damage response process could result in the accumulation of DNA damage.
Prior research has shown an increase in H2AX phosphorylation, suggesting a possible accumulation of DNA damage, in people with neurodegenerative diseases, such as PD (Abugable et al., 2019; Gonzalez-Hunt & Sanders, 2020). Although the exact nature of this increase in PD patients is unknown, past research with ATM and synucleinopathy models of PD suggests that defective DNA damage repair pathways could be a possible cause (Milanese et al., 2018). Additionally, research into alpha-synuclein and DNA damage shows an increase in DSBs following removal of alpha-synuclein and implicates alpha-synuclein in the DNA damage response (Schaser et al., 2019).
Overall, this is an important topic to study due to the deleterious outcomes that can result from nuclear DNA damage (Milanese et al., 2018). Because DNA codes for other biological molecules such as RNA and proteins, damage at the DNA level can have long-lasting effects. These effects are exacerbated in neurons and other similar cells because these cells do not replicate (Milanese et al., 2018).
The Current Study
Prior research shows an increase in H2AX phosphorylation in PD models involving alpha-synuclein, suggesting the possible accumulation of double-stranded DNA breaks in PD (Milanese et al., 2018). Research also shows an increase in ATM activation in synucleinopathy models of PD, indicating that the increase in DNA damage could be related to alterations in the DNA damage
response (Milanese et al., 2018). The current study aimed to explore these findings within LRRK2 G2019S models of PD. The hypothesis of this study was as follows: the implication of ATM and LRRK2 activation in LRRK2 G2019S PD indicates a relationship between a DNA damage response through ATM, LRRK2 kinase activity, and the accumulation of DNA damage.
Methods Cell Culture and Treatment
The experiments of this study were conducted using lymphoblastoid cell lines (LCLs) obtained from the Coriell Institute for Medical Research Biobank (Table 1). Two lines each of healthy control (ND00011 and ND03000) and LRRK2 G2019S (ND00264 and ND01277) cells were grown in RPMI-1640 cell growth media with 15% fetal bovine serum (FBS). Once every 3-4 days, the cells were passaged.
ATM kinase activity was inhibited through treatment with KU-60019 (Selleck Chemicals) at a dose of 1 M. LRRK2 kinase activity was inhibited through treatment with MLi-2 (kindly gifted by Dr. Andrew West, Duke University) at a dose of 100nM. Following a 24 hour treatment, the cells were plated for ICC, as per the protocol described below.
Western Blotting
Protein was collected from 5 million cells in 100 L lysis buffer solution: RIPA buffer (Sigma), 50 L /mL protease inhibitor cocktail (Sigma), 1X Halt phosphatase inhibitor (Thermo Fisher Scientific). The concentration of this protein was quantified using a DC Assay (Bio-Rad Laboratories) and 75 g of protein sample were added to NuPAGE sample buffer (Thermo Fisher Scientific) and dithiothreitol (reducing agent) in a 7:2:1 ratio (7 sample : 2 sample buffer : 1 reducing agent). The samples were mixed and denatured at 100C for 5 minutes. SDS-PAGE was
conducted on these samples using 4-20% polyacrylamide precasted gels (Bio-Rad Laboratories). Following the electrophoresis, protein was transferred from the gels to nitrocellulose membranes using the Trans-Blot Turbo system (Bio-Rad Laboratories). Non-fat dry milk in phosphate buffered saline (PBS)-Tween was used to block the blots for 30 minutes. The blots were then incubated overnight with primary antibodies (ATM pS1981 Abcam ab81292 at 1:10,000, P53 pS15 Cell Signaling Technologies 9284T at 1:2,000, CHK2 pT68 Cell Signaling Technologies 2197T at 1:2,000, -actin Novus Biologicals NBP1-47423 at 1:20,000).
The next day, the blots were washed with PBS and incubated with IRdye infrared fluorescent secondary antibodies (LI-COR) for 45 minutes at a concentration of 1:10,000. The blots were washed with PBS again, dried, scanned with Odyssey imaging system (Li-COR) and analyzed using the Image Studio Lite software (LI-COR).
Immunocytochemistry (ICC)
Uncoated cover slips were placed in wells of a 24-well plate. Following 3-4 days of growth in RPMI-1640 cell growth media with 15% FBS, cells were plated in PBS at a concentration of 3 million cells/mL, 500 L total per well, and were incubated at room temperature for 30 minutes. Following the incubation, the PBS was removed and the cells were fixed in 4% paraformaldehyde for 10 minutes. The wells were then washed with PBS, placed on a shaker with permeabilization buffer (1X PBS with 5% donkey serum and 0.2% Triton 100-X) for 10 minutes, and blocked with blocking buffer (1X PBS with 5% donkey serum) for 30 minutes on a shaker. Following blocking, the wells were incubated overnight with primary antibodies in blocking/antibody buffer (H2AX Millipore Sigma JBW30 at 1:2,000).
The next day, the wells were washed with PBS and incubated with secondary antibodies in blocking/antibody buffer for 1 hour (Alexa Fluor 594 donkey anti-mouse at 1:500, Thermo Fisher
Scientific). Wells were washed with PBS again and incubated with Hoechst in PBS for 5 minutes (2 g/mL). Following this incubation, the wells were rinsed with distilled water and the cover slips were mounted on slides using Prolong Diamond antifade mounting media (Thermo Fisher Scientific). The slides were visualized using Echo Laboratories Revolve Fluorescent Microscope at 60X magnification. Approximately 100 cells for each trial were imaged, to provide a total number of approximately 300 cells for each treatment condition. The ICC results were quantified through an unblinded process using Fiji ImageJ. The number of H2AX foci per cell was counted as a measure of H2AX phosphorylation and the foci per cell counts were used in the statistical analyses described below.
Statistical Analyses
Statistical analyses were conducted using the GraphPad Prism software. An independent samples t-test was used to study the differences in baseline phosphorylation of H2AX between healthy control and LRRK2 G2019S cells. An independent samples t-test was also used to study the differences in the phosphorylation of ATM and its substrates, P53 and CHK2. A 2-way ANOVA (cell line X treatment condition) was conducted to study the effect of ATM kinase inhibition and LRRK2 kinase inhibition in healthy control and LRRK2 G2019S cells.
Results
Increased phosphorylation of H2AX due to LRRK2 G2019S
The phosphorylation of histone H2AX in healthy control and LRRK2 G2019S cell lines was studied as a proxy for DNA DSBs. Greater H2AX phosphorylation would indirectly suggest the accumulation of DSBs. To study baseline differences in phosphorylation of H2AX between healthy control and LRRK2 G2019S cell lines, an independent samples t-test was conducted. The
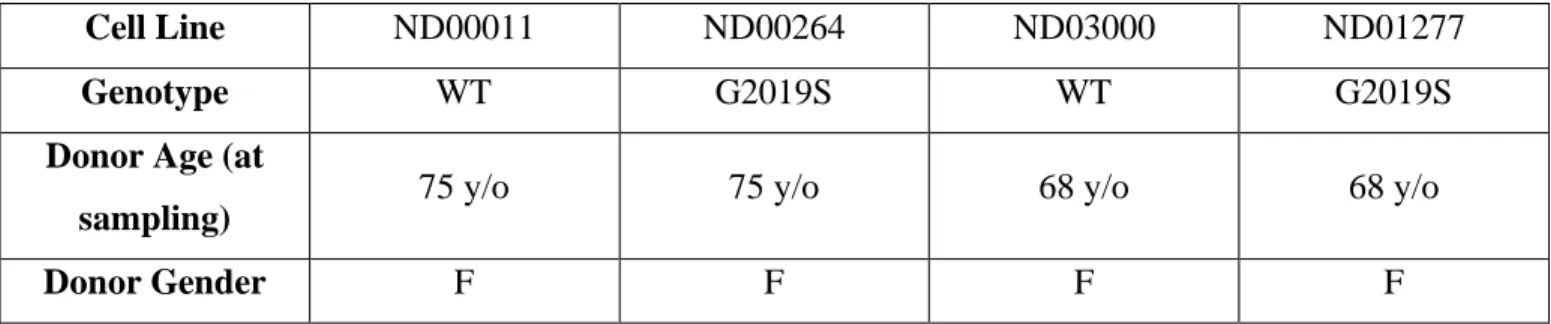
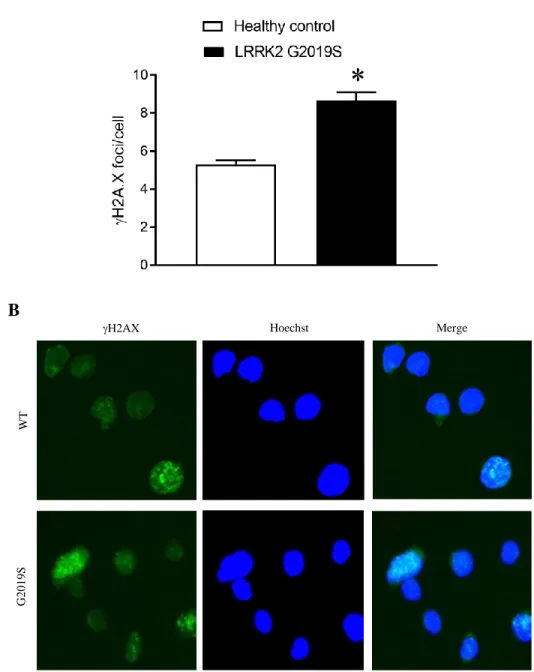
results of the t-test analysis showed significantly greater phosphorylation of H2AX at site Ser139 in LRRK2 G2019S cells when compared to healthy control cells (t(438) = 6.90, p<0.0001). That is, the LRRK2 G2019S cells appeared to have greater DNA damage than the healthy control cells. Figure 2 illustrates these baseline differences.
Activation of ATM kinase due to LRRK2 G2019S
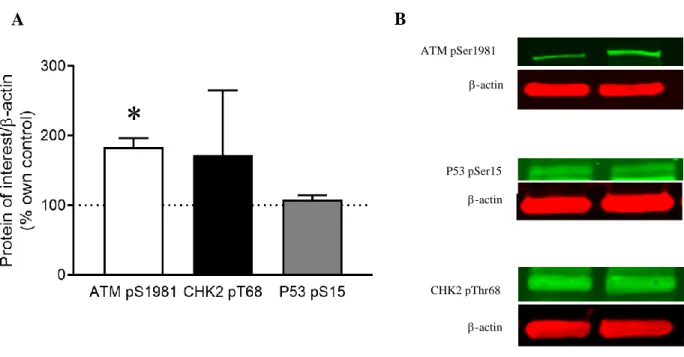
The phosphorylation of ATM was measured in order to evaluate the activation of ATM kinase and, therefore, the possible activation of a DNA damage response pathway involving ATM. To study baseline differences in ATM kinase activation between healthy control and LRRK2 G2019S cell lines, an independent samples t-test was conducted. The results of the t-test analysis showed significantly greater ATM kinase activity at phosphorylation site S1981 in LRRK2 G2019S cells when compared to healthy control cells (t(2) = 6.368, p = 0.012). This indicated a possible increase in the activation of a DNA damage response pathway in LRRK2 G2019S cells than in healthy control cells. Figure 3 illustrates the baseline differences in ATM kinase activation by representing the activation in LRRK2 G2019S cell lines as a percentage normalized to the healthy controls.
No significant differences in P53 and CHK2 activation as a result of LRRK2 G2019S
The activation of P53 and CHK2 was studied in order to determine if the DNA damage response pathway involving ATM was activated (Figure 1). Greater phosphorylation of P53 and CHK2, along with greater phosphorylation of ATM, would suggest that this pathway is activated. To study baseline differences in P53 activation between healthy control and LRRK2 G2019S cell lines, an independent samples t-test was conducted. The results of the t-test analysis indicated no significant baseline differences in P53 activation at phosphorylation site S15 (t(2) = 1.231, p = 0.172). To study baseline differences in CHK2 activation between healthy control and PD cell
lines, an independent samples t-test was conducted. The results of the t-test indicated no significant baseline differences in CHK2 activation at phosphorylation site T68 (t(2) = 0.778, p = 0.259). In sum, although there was greater ATM phosphorylation in LRRK2 G2019S cells, there was no significant difference in P53 or CHK2 phosphorylation between LRRK2 G2019S and healthy control cells. Figure 3 illustrates the results of these P53 and CHK2 activation t-tests by representing the activation in PD cell lines as a percentage normalized to healthy controls.
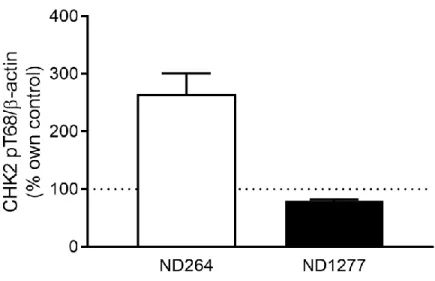
CHK2 activation appeared to differ based on the specific PD cell line, with an apparent increase in CHK2 activation in ND264 and an apparent decrease in CHK2 activation in ND1277. Taken together, there did not appear to be a significant difference in CHK2 phosphorylation between LRRK2 G2019S and healthy control cells. Figure 4 shows this difference in CHK2 activation between the two different PD cell lines.
Inhibition of ATM kinase activity results in reversal of increased H2AX phosphorylation related to LRRK2 G2019S
Phosphorylation of H2AX following inhibition of ATM kinase activity was studied to determine if there is a possible relationship between a DNA damage response pathway through ATM and the accumulation of DNA damage in LRRK2 G2019S cells. A decrease in H2AX phosphorylation following inhibition of ATM kinase activity would suggest that such a relationship might exist. To study the effect of ATM kinase inhibitor KU-60019 on H2AX phosphorylation, a two-way ANOVA was conducted. The results of this two-way ANOVA analysis revealed a significant main effect of treatment condition on H2AX phosphorylation, with a decrease in H2AX phosphorylation in cells treated with KU-600019 (F(1, 2312) = 37.58, p<0.001). The two-way ANOVA analysis also revealed a significant main effect of cell line on H2AX phosphorylation, with lower levels of H2AX phosphorylation in the healthy control cells
than the LRRK2 G2019S cells (F(1, 2312) = 29.50, p<0.001). Finally, this two-way ANOVA analysis showed a significant interaction between treatment condition and cell line (F(1, 2312) = 4.85, p = 0.0277). These results indicated that the increased accumulation of DNA damage in LRRK2 G2019S cells was reversed when ATM kinase activity was inhibited. Figure 5 illustrates these differences in H2AX phosphorylation as a measure of H2AX foci per cell.
Inhibition of LRRK2 kinase activity results in reversal of increased H2AX phosphorylation related to LRRK2 G2019S
Phosphorylation of H2AX following inhibition of LRRK2 kinase activity was measured to determine if LRRK2 kinase could be involved in the accumulation of DNA damage in LRRK2 G2019S cells. A decrease in H2AX phosphorylation following inhibition of LRRK2 kinase activity would suggest that LRRK2 kinase is somehow involved in this DNA damage accumulation. To study the effect of LRRK2 kinase inhibitor MLi-2 on H2AX phosphorylation, a two-way ANOVA was conducted. The results of this two-way ANOVA analysis showed a significant main effect of treatment condition on H2AX phosphorylation, with a decrease in H2AX phosphorylation in cells treated with MLi-2 (F(1, 2408) = 40.60, p<0.0001). This two-way ANOVA analysis also showed a significant main effect of cell line on H2AX phosphorylation, with lower levels of H2AX phosphorylation in the healthy control cells than the PD cells (F(1, 2408) = 29.13, p<0.001). Finally, the two-way ANOVA analysis revealed a significant interaction between treatment condition and cell line (F(1, 2408) = 6.845, p = 0.0089). These results showed that the increased accumulation of DNA damage in LRRK2 G2019S cells was reversed when LRRK2 kinase activity was inhibited. Figure 6 illustrates these differences in H2AX phosphorylation as a measure of H2AX foci per cell.
Discussion
The goal of this study was to evaluate the role of ataxia telangiectasia mutated (ATM), a protein involved in DNA damage response, and leucine rich repeat kinase 2 (LRRK2), a kinase protein, in the accumulation of DNA damage in PD. I hypothesized that the implication of ATM and LRRK2 activation in LRRK2 G2019S PD indicates a relationship between a DNA damage response through ATM, LRRK2 kinase activity, and the accumulation of DNA damage. The results of this study partially supported the hypothesis that a DNA damage response process through ATM is implicated in the greater DNA damage present in LRRK2 G2019S PD. Additionally, the results supported the hypothesis that LRRK2 kinase activity is related to the accumulation of DNA damage in PD.
As predicted, the results showed that baseline levels of DNA damage are greater in LRRK2 G2019S cell lines than in healthy controls. More specifically, the use of H2AX in measuring this DNA damage as a proxy for DSBs suggested that this damage might consist of DSBs in DNA. In order to accurately characterize this DNA damage as DSBs, future experiments should involve comet assays, which allow for the specific detection of DSBs (Santoro, Ferraiuolo, Morgano, Muti, & Strano, 2016). The next question concerned the mechanism through which this DNA damage accumulated. To determine if the DNA damage was related to a DNA damage response pathway involving ATM, the phosphorylation/activation of ATM and its substrates P53 and CHK2 was measured in LRRK2 G2019S and healthy control cells. The results showed higher levels of ATM activation in LRRK2 G2019S cell lines than in healthy controls. This suggested that an ATM-mediated DNA damage response pathway is likely related to the DSB accumulation in LRRK2 G2019S cells. More specifically, an alteration in such a pathway could be a possible cause of the increased levels of DNA damage in the PD cells. However, it is also possible that the increased
levels of DNA damage caused the dysregulation in this DNA damage response. Further studies can provide more insight into the direction of this relationship.
The activation of P53 and CHK2, substrates of ATM, would confirm the relationship between an ATM-mediated DNA damage response pathway (Figure 1) and the accumulation of DSBs in LRRK2 G2019S cells. However, the results of this study did not support this hypothesis. There was no apparent difference in the phosphorylation of P53 between the LRRK2 G2019S and healthy control cell lines. Additionally, there was no overall difference in the phosphorylation of CHK2 between the LRRK2 G2019S and healthy control cell lines. However, while the P53 activation was fairly even across PD cell lines, the CHK2 activation appeared to depend on the specific PD cell line. That is, one LRRK2 G2019S cell line (ND00264) expressed greater levels of CHK2 phosphorylation than the healthy controls while the other LRRK2 G2019S cell line (ND01277) expressed lower levels than the healthy controls. When averaged together, the overall effect appeared to show no difference in CHK2 activation between the PD and healthy control cell lines. This difference between PD cell lines might be a result of innate biological differences between the cell samples. Further research including additional LRRK2 G2019S cell lines should clarify this discrepancy.
The fact that there was a significant difference in ATM activation between LRRK2 G2019S and healthy control cells, but no significant difference in P53 or CHK2 activation could have a couple possible explanations. It is possible that ATM actually acts through a different substrate (other than P53 and CHK2). However, it is also possible that this specific phenotype (phosphorylation of substrates of ATM) is weak in LCLs. If there is a problem in the pathway that allows ATM to phosphorylate P53 and CHK2 in LRRK2 G2019S PD, the reversal results seen in this study are a likely outcome. In this case, a DNA damage response pathway involving ATM
might still be related to the accumulation of DSBs, despite the apparent lack of difference in P53 and CHK2 activation between LRRK2 G2019S and healthy control cells. Further research into this topic is required to determine the nature of this relationship between ATM-mediated DNA damage repair and the accumulation of DSBs in PD.
If a DNA damage response pathway via ATM is involved in the greater phosphorylation of H2AX, inhibiting ATM activation should cause a decrease in the number of H2AX foci, indicating a decrease in the amount of DNA damage present. The results of this study confirmed this. When cells were treated with an ATM kinase inhibitor (KU-60019), there was an apparent reversal in DSBs, as measured through H2AX phosphorylation at site Ser139. In fact, the DNA damage in the LRRK2 G2019S cells was reversed back to the baseline levels of DNA damage in the healthy controls (mean baseline foci per cell in healthy controls = 7.70, mean foci per cell in PD cells after treatment with KU-60019 = 7.48). This confirms that alterations in the DNA damage response through ATM in LRRK2 G2019S PD are related to the greater H2AX phosphorylation. When ATM kinase activity is inhibited, the DNA damage is restored back to the normal levels of healthy controls.
This study revealed an interesting association between LRRK2 kinase activity and DNA damage in PD. As with the ATM kinase inhibitor, when cells were treated with a LRRK2 kinase inhibitor (MLi-2), there was an apparent reversal in DNA damage, as measured through H2AX phosphorylation at site Ser139. Again, the DNA damage in the LRRK2 G2019S cells was reversed back to the baseline levels of DNA damage in the healthy controls (mean baseline foci per cell in healthy controls = 7.70, mean foci per cell in PD cells after treatment with MLi-2 = 7.40). Although there is very little prior research to suggest that LRRK2 is related to DNA damage, these results implicated LRRK2 kinase activity in the increased DNA damage in PD. The results of this study
suggested that, in addition to the increased ATM kinase activity, increased LRRK2 kinase activity likely also contributes to the accumulation of DNA damage in LRRK2 G2019S PD. This DNA damage might be the result of a dysregulated DNA damage response, but the cause of the increase in DNA damage is still unknown. Additionally, as of now, the exact nature of the possible relationship between LRRK2 kinase activity and the ATM-mediated DNA damage response pathway is unknown. Because manipulation of LRRK2 kinase activity resulted in a decrease in H2AX phosphorylation, it is possible that ATM is downstream of LRRK2 in the DNA damage response pathway. If this were the case, alterations in LRRK2 would affect ATM, which would alter the whole DNA damage response pathway and ultimately result in greater DNA damage. Further research should evaluate the potential role of LRRK2 in this pathway.
Accumulation of DNA damage can cause deleterious effects in biological molecules within cells, especially within non-replicating cells such neurons. As such, it is important to conduct research into the mechanisms through which DNA damage accumulates. The results of this study provide novel insight into the implication of ATM and LRRK2 in the accumulation of DNA damage in PD. Although prior research suggests some link between DSBs in PD and DNA damage response pathways involving ATM, there is very little research that implicates LRRK2 in this process. The consideration of LRRK2 in the accumulation of DNA damage and the DNA damage response process can open doors to further research into the role of nuclear DNA damage in PD and, eventually, possible therapeutic targets for PD. In the future, the exact role of LRRK2 in DNA damage accumulation as well as the nature of the involvement of LRRK2 in DNA damage response should be studied. Additionally, the role of other DNA damage response pathways in LRRK2 G2019S PD and the possible role of LRRK2 kinase activity in these pathways can be explored. Finally, this study should be expanded to neurons and other cell types.
Conclusion
The results of this study showed that there are inherently higher levels of DNA damage, as measured through H2AX phosphorylation, in LRRK2 G2019S PD cells than in healthy controls. Greater phosphorylation of ATM in LRRK2 G2019S cell lines indicates that a DNA damage response pathway involving ATM might play a role in the difference in DNA damage between PD and healthy control cells. However, implication of an ATM-mediated DNA damage response pathway could not be confirmed in this study due to the lack of a clear difference in the phosphorylation of P53 and CHK2 (two substrates of ATM). The reversal of DNA damage in LRRK2 G2019S cells following inhibition of ATM kinase activity indicates a relationship between a DNA damage pathway involving ATM and DNA damage accumulation in PD cells. Interestingly, the reversal of DNA damage in LRRK2 G2019S cells following inhibition of LRRK2 kinase activity suggests that abnormalities in the kinase function of LRRK2 might be related to the accumulation of DNA damage in PD cells as well.
References
Abugable, A. A., Morris, J. L. M., Palminha, N. M., Zaksauskaite, R., Ray, S., & El-Khamisy, S. F. (2019). DNA repair and neurological disease: From molecular understanding to the development of diagnostics and model organisms. DNA Repair (Amst), 81, 102669. doi:10.1016/j.dnarep.2019.102669
Beitz, J. M. (2014). Parkinson's disease: a review. Front Biosci (Schol Ed), 6, 65-74. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/24389262
Berquist, B. R., & Wilson, D. M., 3rd. (2012). Pathways for repairing and tolerating the spectrum of oxidative DNA lesions. Cancer Lett, 327(1-2), 61-72. doi:10.1016/j.canlet.2012.02.001 Bradbury, J. M., & Jackson, S. P. (2003). ATM and ATR. Curr Biol, 13(12), R468.
doi:10.1016/s0960-9822(03)00403-2
Chen, Z., Cao, Z., Zhang, W., Gu, M., Zhou, Z. D., Li, B., . . . Zeng, L. (2017). LRRK2 interacts with ATM and regulates Mdm2-p53 cell proliferation axis in response to genotoxic stress. Hum Mol Genet, 26(22), 4494-4505. doi:10.1093/hmg/ddx337
Eilam, R., Peter, Y., Groner, Y., & Segal, M. (2003). Late degeneration of nigro-striatal neurons in ATM-/- mice. Neuroscience, 121(1), 83-98. doi:10.1016/s0306-4522(03)00322-1 Gonzalez-Hunt, C. P., & Sanders, L. H. (2020). DNA damage and repair in Parkinson's disease:
Recent advances and new opportunities. J Neurosci Res. doi:10.1002/jnr.24592
Hardy, J., Cai, H., Cookson, M. R., Gwinn-Hardy, K., & Singleton, A. (2006). Genetics of Parkinson's disease and parkinsonism. Ann Neurol, 60(4), 389-398. doi:10.1002/ana.21022 Hernandez, D., Paisan Ruiz, C., Crawley, A., Malkani, R., Werner, J., Gwinn-Hardy, K., . . . Singleton, A. (2005). The dardarin G 2019 S mutation is a common cause of Parkinson's disease but not other neurodegenerative diseases. Neurosci Lett, 389(3), 137-139. doi:10.1016/j.neulet.2005.07.044
Hernandez, D. G., Reed, X., & Singleton, A. B. (2016). Genetics in Parkinson disease: Mendelian versus non-Mendelian inheritance. J Neurochem, 139 Suppl 1, 59-74. doi:10.1111/jnc.13593
Khanna, K. K. (2000). Cancer risk and the ATM gene: a continuing debate. J Natl Cancer Inst, 92(10), 795-802. doi:10.1093/jnci/92.10.795
Li, J. Q., Tan, L., & Yu, J. T. (2014). The role of the LRRK2 gene in Parkinsonism. Mol Neurodegener, 9, 47. doi:10.1186/1750-1326-9-47
Luzon-Toro, B., Rubio de la Torre, E., Delgado, A., Perez-Tur, J., & Hilfiker, S. (2007). Mechanistic insight into the dominant mode of the Parkinson's disease-associated G2019S LRRK2 mutation. Hum Mol Genet, 16(17), 2031-2039. doi:10.1093/hmg/ddm151
McKinnon, P. J. (2013). Maintaining genome stability in the nervous system. Nat Neurosci, 16(11), 1523-1529. doi:10.1038/nn.3537
Milanese, C., Cerri, S., Ulusoy, A., Gornati, S. V., Plat, A., Gabriels, S., . . . Mastroberardino, P. G. (2018). Activation of the DNA damage response in vivo in synucleinopathy models of Parkinson's disease. Cell Death Dis, 9(8), 818. doi:10.1038/s41419-018-0848-7
Paull, T. T. (2015). Mechanisms of ATM Activation. Annu Rev Biochem, 84, 711-738. doi:10.1146/annurev-biochem-060614-034335
Podhorecka, M., Skladanowski, A., & Bozko, P. (2010). H2AX Phosphorylation: Its Role in DNA Damage Response and Cancer Therapy. J Nucleic Acids, 2010. doi:10.4061/2010/920161 Roos, W. P., & Kaina, B. (2006). DNA damage-induced cell death by apoptosis. Trends Mol Med,
12(9), 440-450. doi:10.1016/j.molmed.2006.07.007
Santoro, R., Ferraiuolo, M., Morgano, G. P., Muti, P., & Strano, S. (2016). Comet Assay in Cancer Chemoprevention. Methods Mol Biol, 1379, 99-105. doi:10.1007/978-1-4939-3191-0_9 Schaser, A. J., Osterberg, V. R., Dent, S. E., Stackhouse, T. L., Wakeham, C. M., Boutros, S. W.,
. . . Unni, V. K. (2019). Alpha-synuclein is a DNA binding protein that modulates DNA repair with implications for Lewy body disorders. Sci Rep, 9(1), 10919. doi:10.1038/s41598-019-47227-z
Shiloh, Y. (2003). ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer, 3(3), 155-168. doi:10.1038/nrc1011
Sulli, G., Di Micco, R., & d'Adda di Fagagna, F. (2012). Crosstalk between chromatin state and DNA damage response in cellular senescence and cancer. Nat Rev Cancer, 12(10), 709-720. doi:10.1038/nrc3344
Welty, S., Teng, Y., Liang, Z., Zhao, W., Sanders, L. H., Greenamyre, J. T., . . . Lan, L. (2018). RAD52 is required for RNA-templated recombination repair in post-mitotic neurons. J Biol Chem, 293(4), 1353-1362. doi:10.1074/jbc.M117.808402
West, A. B., Moore, D. J., Biskup, S., Bugayenko, A., Smith, W. W., Ross, C. A., . . . Dawson, T. M. (2005). Parkinson's disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci U S A, 102(46), 16842-16847. doi:10.1073/pnas.0507360102
Appendix
Cell Line ND00011 ND00264 ND03000 ND01277
Genotype WT G2019S WT G2019S
Donor Age (at
sampling) 75 y/o 75 y/o 68 y/o 68 y/o
Donor Gender F F F F
Table 1. Information regarding genotypes and donor demographics for each lymphoblastoid cell line used. Information was collected from Coriell Institute database.
Figure 1. Schematic of DNA damage response pathways (Sulli, Di Micco, & d'Adda di Fagagna, 2012). The pathway of interest in the current study involves ATM, CHK2, and P53.
W T Merge Hoechst H2AX G 2019S
Figure 2. Baseline H2AX phosphorylation in healthy control and LRRK2 G2019S cell lines. A) An independent samples t-test revealed significant differences in H2AX phosphorylation at Ser139 between healthy control (n=2) and LRRK2 G2019S (n=2) cell lines (p<0.001). B) A fluorescent microscope was used to image H2AX phosphorylation as foci.
A
Figure 3. ATM pS1981, CHK2 pT68, and P53 pS15 phosphorylation in LRRK2 G2019S cell lines normalized to -actin. A) Protein phosphorylation was quantified as a percent control measure. Activation of proteins in healthy control cell lines is marked by the dashed line at 100%. An independent samples t-test revealed a significant difference in phosphorylation of ATM at S1981 between healthy control (n=2) and LRRK2 G2019S (n=2) cell lines (p=0.012). Independent samples t-tests did not yield significant results for differences between groups in P53 and CHK2. B) Odyssey Imaging System was used to visualize phosphorylated proteins of interest.
ATM pSer1981 -actin -actin P53 pSer15 CHK2 pThr68 -actin A B
Figure 4. CHK2 pT68 activation in 2 LRRK2 G2019S cell lines (ND00264 and ND01277) normalized to -actin as a percent control measure. Activation of proteins in healthy control cell lines is marked by the dashed line at 100%.
G 2019S D M S O W T D M S O W T K U G 2019S K U Merge Hoechst H2AX
Figure 5. H2AX phosphorylation in healthy control and LRRK2 G2019S cell lines following treatment with DMSO and ATM kinase inhibitor KU-60019. A) A two-way ANOVA revealed a significant difference in H2AX phosphorylation after treatment with KU-60019 (p<0.001). B) A fluorescent microscope was used to image H2AX phosphorylation as foci.
A
G 2019S D M S O W T D M S O Merge Hoechst H2AX W T M L i-2 G 2019S M L i -2
Figure 6. H2AX phosphorylation in healthy control and LRRK2 G2019S cell lines following treatment with DMSO and LRRK2 kinase inhibitor MLi-2. A) A two-way ANOVA revealed a significant difference in H2AX phosphorylation after treatment with MLi-2 (p<0.001). B) A fluorescent microscope was used to image H2AX as foci.
A