systemic autoimmunity in murine models
Xuezhi Dai, … , Jane H. Buckner, David J. Rawlings
J Clin Invest.
2013;
123(5)
:2024-2036.
https://doi.org/10.1172/JCI66963
.
Multiple autoimmune diseases, including type 1 diabetes, rheumatoid arthritis, Graves
disease, and systemic lupus erythematosus, are associated with an allelic variant of protein
tyrosine phosphatase nonreceptor 22 (
PTPN22
), which encodes the protein LYP. To model
the human disease-linked variant LYP-R620W, we generated knockin mice expressing the
analogous mutation, R619W, in the murine ortholog PEST domain phosphatase (PEP). In
contrast with a previous report, we found that this variant exhibits normal protein stability,
but significantly alters lymphocyte function. Aged knockin mice exhibited effector T cell
expansion and transitional, germinal center, and age-related B cell expansion as well as the
development of autoantibodies and systemic autoimmunity. Further, PEP-R619W affected B
cell selection and B lineage–restricted variant expression and was sufficient to promote
autoimmunity. Consistent with these features, PEP-R619W lymphocytes were
hyperresponsive to antigen-receptor engagement with a distinct profile of
tyrosine-phosphorylated substrates. Thus, PEP-R619W uniquely modulates T and B cell
homeostasis, leading to a loss in tolerance and autoimmunity.
Research Article
Autoimmunity
Find the latest version:
A disease-associated PTPN22
variant promotes systemic
autoimmunity in murine models
Xuezhi Dai,1,2 Richard G. James,3 Tania Habib,4 Swati Singh,1,2 Shaun Jackson,1,2
Socheath Khim,1,2 Randall T. Moon,3,5 Denny Liggitt,6
Alejandro Wolf-Yadlin,7 Jane H. Buckner,4 and David J. Rawlings1,2,8
1Department of Pediatrics, University of Washington School of Medicine, Seattle, Washington, USA. 2Seattle Children’s Research Institute,
Seattle, Washington, USA. 3Department of Pharmacology, University of Washington School of Medicine,
Seattle, Washington, USA. 4Translational Research Program, Benaroya Research Institute, Seattle, Washington, USA. 5Howard Hughes Medical Institute, 6Department of Comparative Medicine, 7Department of Genome Sciences, and
8Department of Immunology, University of Washington School of Medicine, Seattle, Washington, USA.
Multiple autoimmune diseases, including type 1 diabetes, rheumatoid arthritis, Graves disease, and systemic
lupus erythematosus, are associated with an allelic variant of protein tyrosine phosphatase nonreceptor 22
(PTPN22), which encodes the protein LYP. To model the human disease-linked variant LYP-R620W, we
generated knockin mice expressing the analogous mutation, R619W, in the murine ortholog PEST domain
phosphatase (PEP). In contrast with a previous report, we found that this variant exhibits normal protein
stability, but significantly alters lymphocyte function. Aged knockin mice exhibited effector T cell expansion
and transitional, germinal center, and age-related B cell expansion as well as the development of
autoanti-bodies and systemic autoimmunity. Further, PEP-R619W affected B cell selection and B lineage–restricted
variant expression and was sufficient to promote autoimmunity. Consistent with these features, PEP-R619W
lymphocytes were hyperresponsive to antigen-receptor engagement with a distinct profile of
tyrosine-phos-phorylated substrates. Thus, PEP-R619W uniquely modulates T and B cell homeostasis, leading to a loss in
tolerance and autoimmunity.
Introduction
An allelic variant of protein tyrosine phosphatase nonreceptor 22 (PTPN22) is strongly associated with multiple autoimmune diseases including type 1 diabetes (T1D), SLE, RA, Graves disease, and others (1–4). The risk allele comprises a single-nucleotide polymorphism, C1858T, resulting in an amino acid change from arginine to tryp-tophan at position 620 (R620W) of the encoded protein LYP (5). While LYP functions as a negative regulator in TCR signaling (6, 7), the precise mechanism or mechanisms whereby LYP-R620W contrib-utes to autoimmunity remain unclear. The variant allele alters the first of 4 carboxyterminal, proline-rich regions of LYP, leading to a disruption in its association with Csk, a negative regulator of src-family kinases. Work to date, however, has led to contradictory con-clusions with respect to the functional activity of LYP-R620W, with data supporting its role as either a gain- or a loss-of-function variant in antigen-receptor (AR) signaling (5, 8–11). For example, overexpres-sion of LYP-R620W inhibits AR-dependent calcium flux in T or B cell lines (5, 9). Primary T and B cells from healthy individuals with the variant allele exhibit deficits in AR-induced calcium mobilization (8, 9, 12). In contrast, overexpression of mutant LYP, in conjunction with Csk, promotes increased calcium flux in T cell lines (10), and primary T and B cells from RA subjects carrying the risk allele exhibit augmented MAPK signaling in response to AR engagement (11).
The murine ortholog of Ptpn22 encodes the protein PEST domain phosphatase (PEP). Targeted disruption of Ptpn22
results in modest, lineage-specific alterations in murine immune
responses (13). AR-induced signaling is enhanced in mem-ory/effector T cells derived from PEP-deficient animals, but unchanged in naive T or B cells. Aged PEP-deficient mice have increased memory/effector T cells and spontaneous GC produc-tion. However, these animals lack autoantibodies or other evi-dence for systemic autoimmunity. Interestingly, crossbreeding of PEP-deficient mice with animals that express an activating mutation within CD45 promotes SLE-like disease (10). Recently, Zhang et al. generated knockin mice expressing PEP-R619W, a mutant analogous to LYP-R620W (11). Similar to PEP-deficient mice, PEP-R619W knockin mice had enlarged spleens and thy-muses, expanded memory/effector T cells, and lymphocyte and dendritic cell hyperresponsiveness. Notably, these authors found a dramatic reduction in PEP protein levels in these mice and also found that both PEP-R619W and LYP-R620W exhibit shortened protein half-lives and increased sensitivity to calpain-mediated degradation in vitro. Of note, knockin mice in the C57BL/6J genetic background did not develop autoimmunity (11).
In the current study, we independently generated PEP-R619W knockin mice. Interestingly, while some of the features of these mice corresponded with those described by Zhang et al. (e.g., expanded memory/effector T cells and lymphocyte hyperresponsiveness), we report some important differences. For example, we found both PEP-R619W and LYP-R620W exhibit normal protein stability in vivo and in vitro. Additionally, although young mice appeared normal, aged heterozygous and homozygous knockin mice (referred to here as T/C and T/T animals, respectively) on a mixed C57BL/6J and 129/Sv genetic background developed characteristics of autoimmunity including a broad range of high-titer autoantibodies and vasculitis,
lymphoid infiltrates in multiple organs, and increased mortality. In addition, T/C and T/T mice, compared with WT littermates, were more susceptible to streptozotocin-induced (STZ-induced) T1D. The PEP variant also affected early B cell homeostasis, leading to an increase in transitional B cells and an enrichment of self-reactive cells, and expression of the variant in B cells alone was sufficient to promote autoimmune disease. Phosphoproteome analysis revealed quantitative differences in tyrosine-phosphorylated substrates downstream of the AR in knockin versus WT or PEP-deficient pri-mary lymphocytes. Together, these results demonstrate that altered lymphocyte selection, function, and homeostasis in PEP-R619W knockin mice leads to a progressive loss in T and B cell tolerance and promotes systemic autoimmunity in a mixed genetic background.
Results
The PTPN22 variant does not impair protein stability. To understand how the PTPN22 variant contributes to autoimmunity, we gen-erated knockin mice with an equivalent mutation targeting
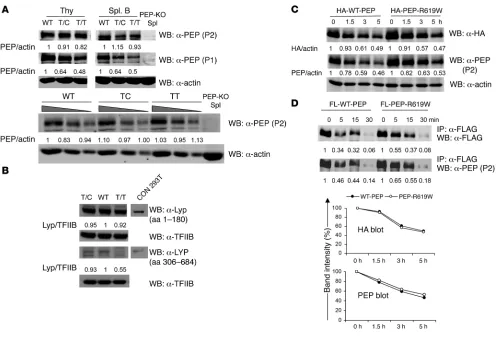
endogenous Ptpn22, leading to expression of the variant protein PEP-R619W (Supplemental Figure 1A; supplemental material available online with this article; doi:10.1172/JCI66963DS1). Presence of the mutation was confirmed by RT-PCR followed by sequencing of cDNA from thymocytes of heterozygous (T/C) or homozygous (T/T) mice (Supplemental Figure 1D). To assess protein expression in T/C, T/T, or WT littermates, Western blot-ting was performed using lysates from thymocytes or purified splenic B cells and 2 alternative anti-PEP Abs (P1 and P2) spe-cific for PEP proline-rich regions 1 and 2, respectively (13). The R619W variant is located within proline-rich region 1. Thus, when using the P1 antibody, T/C and T/T cells showed a pro-gressive decline in relative PEP protein reactivity. In contrast, P2 immunoblots showed comparable PEP expression in all geno-types (Figure 1A). Compared with naive cells, PEP is upregulated in CpG-stimulated B cells and in vitro–generated effector T cells (Supplemental Figure 2, A and C). Again, control and knockin cells showed comparable protein expression (Supplemental
Fig-Figure 1
PEP-R619W and LYP-R620W exhibit no deficit in protein stability. (A) PEP expression in thymocytes and splenic B cells from T/C, T/T, and WT littermates. Cell lysates were subjected to Western blot using anti–PEP-P1, anti–PEP-P2, or anti-actin antibodies. Lower panel shows serial dilution of thymocyte lysates. (B) LYP expression in purified CD3+ T cells from healthy T/T or T/C or control (C/C) subjects. Lysates were blotted
[image:3.585.46.543.79.416.2]ure 2, B and D). Together, these data suggest that the knockin variant alters P1 antibody–binding affinity and, possibly, the affinity of the mutant protein for candidate interacting proteins, but has little or no impact on PEP expression.
As these findings appeared to differ from a recent report (11), we next determined whether the LYP-R620W exhibited evi-dence for altered expression in primary human cells. We blotted lysates derived from CD3+ T cells isolated from healthy donors
(with or without the variant allele) using anti-LYP antibodies raised against either the N- (aa 1–180) or the C-terminal regions (aa 306–684; containing the variant residue and utilized in the previous study; ref. 11). Comparable LYP expression was observed in all genotypes using the N-terminal antibody. In con-trast, the signal was decreased in variant cells using the C-termi-nal reagent (Figure 1B and Supplemental Figure 3A). These data indicate that both PEP-R619W and LYP-R620W are expressed at levels equivalent to the WT protein in primary murine and human lymphocytes and suggest that the reported decrease in mutant protein levels may reflect use of reagents affected by rel-ative affinity for WT versus mutant epitopes.
To directly address whether the variant affects PEP protein half-life, Ramos B cells overexpressing HA-tagged WT-PEP versus PEP-R619W were treated with the protein synthesis inhibitor cyclo-heximide (CHX) and protein half-life assessed using anti-HA or anti-PEP P2 antibodies. As shown in Figure 1C, PEP-R619W showed a protein degradation rate nearly identical to that of WT-PEP. Similar results were obtained using Jurkat T cells overexpressing FLAG-tagged PEP versus PEP-R619W and in ex vivo–cultured pri-mary B and T cells derived from WT and T/T mice (Supplemen-tal Figure 2, C and F). Moreover, LYP and LYP-R620W exhibited an equivalent protein half-life in both Ramos B and Jurkat T cells (Supplemental Figure 3B and data not shown).
To assess whether PEP-R619W is more susceptible to proteolysis, overexpressed, FLAG-tagged PEP versus PEP-R619W were immu-noprecipitated from Ramos B cells and subjected to an in vitro calpain 1 cleavage assay. The WT and mutant proteins had similar
cleavage kinetics (Figure 1D). WT-LYP and LYP-R620W also had equivalent cleavage kinetics (Supplemental Figure 3C). Thus, the variant has minimal impact on calpain 1–mediated proteolysis. These combined experiments support the notion that neither the PEP-R619W nor the LYP-R620W variant exhibits impaired pro-tein stability, implying that autoimmunity in subjects expressing LYP-R620W is not simply due to reduced phosphatase expression.
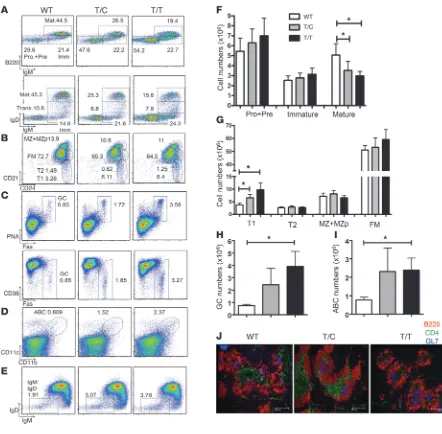
Expression of PEP-R619W promotes autoimmunity in a mixed genetic background. The lymphoid organs appeared normal in young PEP knockin mice (2 to 3 months of age), but spleen size and total cellu-larity were modestly increased in aged T/C and markedly increased in aged T/T mice compared with WT littermates (6 months; Figure 2A). Thymuses from aged T/T mice also contained increased numbers of CD4–CD8– cells comprising primarily B220+CD19+
B cells (Figure 2B and Supplemental Figure 4A). In addition to B cells with a naive-like phenotype (IgM+IgD+), this
popula-tion contained sizable cellular subsets with features of activated (CD5loIgM+), GC (CD38–FAS+), and isotype-switched (IgM–IgD–)
B cells (Supplemental Figure 4B), suggesting ongoing activation within the thymus. Consistent with this idea and previous data (14), purified thymic B cells from aged PEP-R619W knockin mice spontaneously produced antinuclear antibodies (ANA) (Supple-mental Figure 4C). Interestingly, while the presence of CD5+ B cells
in knockin animals differed from studies in NOD mice (15), this population lacked the peritoneal B1 marker, CD11b, expressed on thymic B cells in aged lupus-prone mice (16, 17).
To determine whether the PEP-R619W variant induces autoan-tibody production, we first measured autoantibodies to dsDNA in the serum of T/C, T/T, and WT littermates. At 6 months of age in both males and females, T/C and T/T mice had higher anti-dsDNA autoantibodies than WT littermates (Figure 2C). Moreover, antigen array studies revealed the presence of a broad range of auto-antibodies in aged T/C or T/T mice (Supplemental Figure 5A).
PEP-R619W knockin mice exhibited a modest, but statistically significant, reduction in life span relative to WT mice (Supplemen-tal Figure 6). Histopathology analysis at 6 months of age revealed significant changes, including vasculitis, lymphoid infiltrates, and other changes in nearly all T/T and a proportion of T/C mice, but not WT littermates (Figure 2D and Supplemental Table 1). Lesions were most prominent in the lung and salivary glands, with inter-mittent involvement of other sites including the heart, common bile and pancreatic ducts, intestine, and adjacent mesentery. Renal histology in aged T/C or T/T mice (10 months) revealed renal hypertrophy with glomerular lesions including increased cellular-ity and mesangial matrix, intermittent mesangiolysis or ischemic glomeruli, and infiltration of Mac-2+ cells (Supplemental Figure 7).
Neither T/C nor T/T mice developed evidence of T1D spontane-ously (up to 10 months, data not shown). However, aged T/T mice had significantly increased anti-insulin autoantibodies compared with WT littermates (Figure 2E). To directly assess whether vari-ant expression might contribute to T1D, mice were treated with multiple doses of STZ, a cytotoxic agent that targets insulin-pro-ducing pancreatic β cells (18). As shown in Figure 2F, the incidence of STZ-induced T1D was significantly higher in T/C or T/T mice, relative to WT littermates. Taken together, PEP-R619W induced multiple features of autoimmunity and increased the susceptibil-ity of mice to STZ-induced T1D.
PEP-R619W enhances both thymic-positive and -negative selection. Thy-mocyte numbers and development were comparable among young WT, T/C, and T/T mice (6 to 8 weeks, Supplemental Figure 8A).
Figure 2
Expression of the PEP variant promotes autoimmunity in a mixed genetic background. (A) Increased spleen size and cellularity in 6-month-old T/C and T/T mice. Error bars represent SD based on 8 animals/genotype. **P < 0.01. (B) Infiltration of thymus with B cells in aged T/C and T/T animals. Analysis of total thymocytes (top) and dou-ble-negative (CD4–CD8–) thymocytes (bottom) are shown with
Surface CD5, CD69, and TCRβ are upregulated in double-positive thymocytes (DP; CD4+CD8+) after undergoing positive selection
through the TCR (19). Consistent with enhanced selection, “selec-tion” (TCRβ+CD5+ or TCRβ+CD69+) DP were increased in T/C
and T/T mice (Supplemental Figure 8, B and C). To further test this notion, we crossed knockin mice with OT-II mice, expressing a Vα2/Vβ5 TCR specific for OVA peptide in the context of MHC class II I-Ab (20). Thymic cellularity in OT-II/PEP-R619W T/T
mice was significantly increased when compared with OT-II/WT littermates (Supplemental Figure 8, D and E). To assess whether
PEP-R619W affects thymic negative selection, PEP-R619W knockin mice were bred with HY-TCR trans-genic mice, expressing a TCR recognizing the male-specific antigen HY in the context of MHC class I. As shown in Supplemental Figure 8F, male HY-T/T mice exhib-ited near complete depletion of DP cells consistent with increased negative selection, whereas female HY-T/T mice displayed an increase in CD8+ T cells consistent with
increased positive selection. Thus, PEP-R619W variant enhances both thymic-posi-tive and -negathymic-posi-tive selection.
Expansion of memory/effec-tor T cell compartment in aged knockin mice. Aged PEP-defi-cient mice develop increased numbers of memory/effec-tor T cells, and human subjects harboring the
PTPN22 risk allele also have increased memory T cells (8). FACS analysis revealed slight increases in spleen and LN CD4+ memory/
effector T cells (CD44hi
C-D62Llo) in young T/T mice
(6 to 8 weeks) compared with WT littermates (data not shown). Notably, the percentage and absolute numbers of CD4+
effec-tor/memory T cells were increased in aged T/C and T/T mice compared with WT controls (6 months; Fig-ure 3A). CD4+ T cells in T/T
animals also displayed an activated phenotype, with upregulated CD95, CD69, CD122, and CD80 (Supple-mental Figure 9B). A similar accumulation of activated, memory/effector T cells was present in the CD8+ compartment in
aged T/C and T/T mice (Supplemental Figure 9A).
[image:6.585.48.420.81.452.2]PEP-R619W does not affect Treg development or suppressive activity. As Tregs play a critical role in peripheral T cell tolerance (21) and PEP-deficient mice had increased total Treg numbers (22), we deter-mined whether the variant altered Treg development and/or func-tion. Expression of PEP-R619W did not affect Treg development or absolute numbers in lymphoid organs in young (6 to 8 weeks) mice. However, consistent with increased spleen size, aged T/C and T/T mice had higher absolute Treg numbers (Supplemental Figure 10A).
Figure 3
PEP-R619W alters T cell homeostasis and TCR signaling. (A) Increased CD4+ memory/effector T cells in aged
T/C and T/T versus WT littermates. Splenocytes or LN cells were analyzed by FACS for CD4, CD8, CD44, and CD62L expression. Percentages indicate cells within the CD4+ gate (left panels). Absolute numbers of naive
and memory/effector CD4+ cells (right panel). Error bars represent SD based on 8 animals/genotype. **P < 0.01.
(B) TCR-induced calcium flux in CD4+CD8+ thymocytes (top) and splenic CD4+ T cells (bottom). Cells were
stained with indo-1 AM, anti-CD4, and anti-CD8, then stimulated with biotin-conjugated anti-CD3 antibody and cross-linked by streptavidin (arrow). Ca2+ mobilization was determined by flow cytometry. (C and D) Proliferation
of naive (C) and memory/effector (D) CD4+ T cells. CD4+CD25–CD62Lhi naive T cells and CD4+CD25–CD62Llo/–
Figure 4
PEP-R619W enhances TCR signaling of in vitro–generated effector T cells. (A) TCR-induced calcium flux of in vitro–generated effector T cells. Purified CD4+ T cells from T/T, T/C, and WT mice were stimulated with anti-CD3/CD28 for 3 days and rested in IL-2 medium for 2 days to
generate effector T cells. Ca2+ mobilization was determined by flow cytometry. (B) Enhanced proliferation of T/C and T/T effector T cells. Cells
Tregs from T/T mice inhibited both WT and T/T effector cells as potently as WT Tregs at all target ratios (Supplemental Figure 10B), and WT and T/T Tregs exhibited similar suppressive function across a broad range of anti-CD3 concentrations (data not shown). Of note, T/T-derived effector T cells were equivalently responsive to either WT or T/T Treg–mediated suppression. Thus, autoimmunity in knockin mice is unlikely to be due to either altered Treg development or func-tion or enhanced effector T cell resistance to Treg activity.
PEP-R619W variant enhances TCR signaling in naive and memory T cells. PEP deficiency enhances TCR signaling in effector/memory but not naive T cells (13). To address whether the variant altered TCR responsiveness, we first analyzed TCR-induced calcium flux (23). In contrast with naive PEP-deficient T cells, we observed a gene dose-de-pendent increase in calcium signaling in naive splenic CD4+ T cells
[image:8.585.69.511.79.513.2]derived from T/C and T/T mice (Figure 3B). Similar findings were obtained using naive CD8+ T cells (data not shown), DP thymocytes
Figure 5
Altered B cell homeostasis in aged PEP-R619W knockin mice. (A–E) Representative FACS analysis of B cell populations in various tissues derived from 6-month-old T/C, T/T, and WT littermates. Numbers indicate relative percentages of each developmental population within the B220+ gate. (A) Decreased mature recirculating B cells in BM in knockin mice. BM cells were analyzed for B220, IgM, and IgD expression. (B)
Increased T1 B cells in knockin mice. Splenocytes were stained for B220, CD21, and CD24 expression. Subsets were defined as CD21loCD24hi
(T1), CD21intCD24hi (transitional 2, T2), CD21hiCD24hi (MZ and MZ progenitor; MZ + MZp), and CD21intCD24int (FM) B cells. (C) Increased GC B
cells in knockin mice. Splenocytes were analyzed for B220, PNA, CD38, and FAS expression. (D) Increased ABC in knockin mice. Splenocytes were analyzed for B220, CD11b, and CD11c expression. (E) Increased IgD–IgM– isotype switched B cells in knockin mice. Splenocytes were
(Figure 3B), and in vitro–generated effector T cells (Figure 4A). The enhanced TCR signaling was not due to altered CD3 expression on T cells in PEP-R619W knockin mice (Supplemental Figure 11A).
We next determined whether PEP-R619W affects T cell prolifer-ation. Sort-purified, splenic naive (CD62LhiCD25–), or memory/
effector (CD62Llo/–CD25–) CD4+ T cells were stimulated with
anti-CD3 (or anti-anti-CD3 and anti-CD28) and proliferation quantified
by vital dye dilution. Naive T/C and T/T CD4+ T cells exhibited a
PEP-R619W dose-dependent enhancement in cell proliferation in response to all stimuli (Figure 3C). Similarly, T/C and T/T mem-ory/effector T cells also exhibited increased proliferation relative to WT controls in response to anti-CD3 stimulation (Figure 3D).
Our observations regarding enhanced memory/effector CD4+ T
cell proliferation in T/C and T/T mice differ from data in an
alter-Figure 6
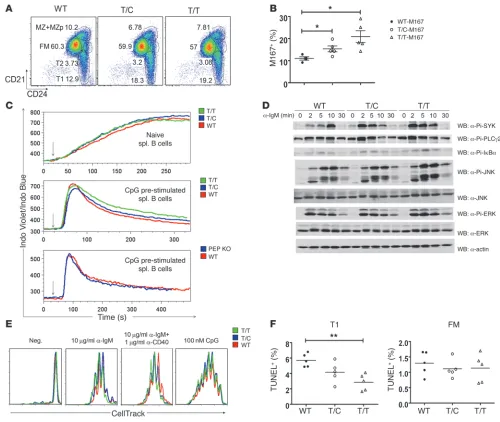
PEP-R619W expression alters B cell selection, augments BCR signaling, and partially protects immature B cells from apoptosis. (A) Increased T1 B cell in 6- to 8-week-old knockin mice. Splenic B cell subsets were defined as in Figure 5. (B) Enhanced selection for M167-Id+ cells in the
MZ compartment in young knockin mice. Splenocytes were stained with anti-B220, anti-CD21, anti-CD24, and anti-M167-Id–specific (where Id, indicates idiotype) antibodies. Percentage of M167-Id+ cells within the MZ gate (CD21hiCD24hiCD23lo/–) is displayed. Each symbol represents an
individual mouse; horizontal bars represent mean ± SEM. *P < 0.05. (C) BCR signaling in naive and CpG prestimulated B cells. Naive B cells (top), CpG prestimulated B cells from T/C, T/T, and WT littermates (middle), and from PEP-deficient and control mice (bottom) were stimulated with anti-IgM antibody at the times indicated by arrows. Induction of Ca2+ mobilization was determined by flow cytometry. (D) PEP-R619W expression
promotes enhanced phosphorylation of BCR-dependent substrates in CpG-prestimulated B cells. CpG-prestimulated B cells were stimulated with anti-IgM antibody for indicated times, and cell lysates were blotted using the indicated antibodies. (E) Proliferation of T/C, T/T, and control B cells. Purified CD43– splenic B cells were labeled with CellTrace and stimulated as indicated; proliferation was determined by CellTrace dilution. (F)
Apoptosis analysis of T1 and FM B cells. Splenocytes were stained with anti-B220, anti-CD21, and anti-CD24. T1 and FM B cells were analyzed for apoptosis by TUNEL. Each symbol represents an individual mouse. *P < 0.05; **P < 0.01. Data shown are representative of 6 (A), 3 (C and
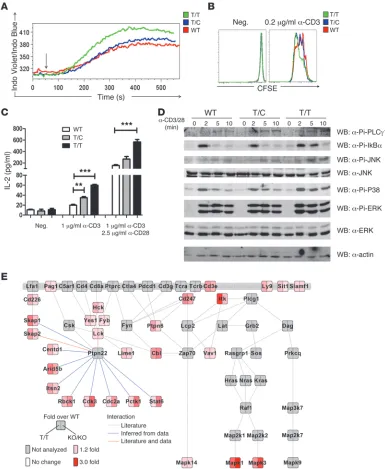
[image:9.585.41.541.80.502.2]native knockin strain describing no functional alteration in this population (11). To clarify this issue, we characterized the func-tions of in vitro–generated effector T cells (13). Upon TCR stimu-lation, calcium flux, T cell proliferation, and IL-2 production were each progressively enhanced in effector T cells derived from T/C and T/T mice compared with controls (Figure 4, A–C).
Next, to directly assess the specific impact of PEP-R619W expres-sion on proximal TCR signaling, we performed detailed biochem-ical analyses using in vitro–generated effector T cells. First, we
analyzed protein phosphorylation by direct Western blotting. Inducible phosphorylations of phospholipase Cγ1 (PLCγ1), IκBα, and MAPKs were enhanced and prolonged in T/T-derived, relative to WT, T cells (Figure 4D).
[image:10.585.43.542.88.494.2]As an unbiased approach to identify alterations in phosphorylated proteins in PEP-R619W or PEP-deficient versus WT T cells, we used isobaric tags for relative and absolute quan-tification (iTRAQ) combined with phospho-tyrosine purification and high-resolution mass spectrometry (24). This approach was
Figure 7
MZ B cells in young knockin mice exhibit a PEP-R619W dose-depen-dent expansion of PC-binding B cells. Thus, PEP-R619W alters B cell repertoire selection and results in enrichment of self-reactive B cells.
Spontaneous GC formation and expansion of GC B cells are hallmarks of many autoimmune-prone mouse strains (34). GC B cells (defined as PNA+FAS+ or CD38–FAS+) were increased in aged
T/C mice, and this change accelerated in T/T animals (6 months; Figure 5, C and H). Consistent with these FACS data, histological analysis revealed larger and more frequent splenic GL7+ GCs in
T/C and T/T mice compared with WT controls (Figure 5J). Age-dependent B cells (ABCs, B220+CD11b+CD11c+) arise at
high frequency in spleens of aged female mice and at earlier time points in lupus-prone animals, and this population is capable of secreting autoantibodies upon stimulation in vitro (35). Interest-ingly, an increased percentage and absolute numbers of ABCs were present in aged T/T compared with WT mice (Figure 5, D and I). In addition, we identified an increased proportion of IgM–IgD–
isotype switched B cells within the spleen of T/C and T/T mice (Figure 5E). Together, PEP-R619W leads to an increase in splenic T1 B cells, a modest decline in BM mature B cells, altered self-reac-tive B cell selection, and a progressive accumulation of GC, ABC, and class-switched B cells in aged T/C and T/T mice.
Altered T1 B cell survival and increased mature B cell activation in knockin mice. The increased T1 B cell compartment in knockin mice prompted us to examine T1 B cell apoptosis in vivo. Apopto-sis in splenic B cells was assessed ex vivo using a TUNEL assay (36). While no changes were observed in FM B cells, T1 B cells from T/T mice had significantly fewer apoptotic cells (Figure 6F).
Next, we evaluated BCR signaling in mature B cell populations. While BCR-triggered calcium flux was comparable in naive B cells from T/T, T/C, and WT mice (Figure 6C), BCR-driven cell prolif-eration was slightly increased by variant expression. B cells from T/C or T/T mice exhibited a consistent increase in proliferation in response to anti-IgM (or anti-IgM plus anti-CD40), but no differ-ences in response to TLR ligands (LPS or CpG; Figure 6E).
Because CpG stimulation leads to upregulation of PEP in B cells (Supplemental Figure 2, A and B), we utilized this approach to mimic PEP levels in early GC B cells as an additional means to assess the impact of PEP-R619W on AR signaling. Anti-IgM–induced calcium flux was slightly enhanced in CpG-prestimulated T/T, relative to WT B cells (Figure 6C). BCR-dependent total protein tyrosine phospho-rylation (pY) and transphosphophospho-rylation of key effectors, including SYK, PLCγ2, IκBα, and JNK, was enhanced and prolonged in T/C and T/T B cells (Supplemental Figure 14 and Figure 6D). In contrast to knockin cells, CpG-prestimulated PEP-deficient B cells exhibited calcium signaling that did not differ from WT cells (Figure 6C), implying that the effect of the R619W allele on AR signaling is dis-tinct from a simple loss of protein expression.
PEP-R619W knockin mice had increased serum levels of IgG1 and IgE (Supplemental Figure 15A). To examine T-independent (TI) antibody responses, we immunized mice with TNP-Ficoll. T/T mice showed increased TNP-specific IgM and IgG3 (Supplemen-tal Figure 15B). Similarly, T-dependent (TD) antibody responses (NP-specific IgM and IgG1) following immunization of mice with NP-chicken γ-globulin (CGG) were elevated in T/T animals (Sup-plemental Figure 15C). Thus, PEP-R619W expression leads to enhanced TI and TD antibody responses.
PEP-R619W expression in B cells is sufficient to promote autoimmunity in a mixed genetic background. To evaluate whether B cell–intrinsic PEP-R619W expression might be sufficient to induce at least some applied concurrently to unstimulated or
anti-CD3/CD28–stim-ulated (5 minutes) in vitro–generated effector T cells from each strain, allowing definitive identification and quantification of 154 distinct phosphotyrosine peptides derived from 80 proteins (Sup-plemental Figure 12A). Detailed analysis of the Ptpn22-regulated phosphoproteome identified peptides corresponding to multiple early components within the TCR pathway, including src-fam-ily kinases, CD247 (Cd3ζ), Itk, Cbl, Skap1/2, Vav1, and others (refs. 25, 26, and Figure 4E). In addition, changes in a number of other proteins with poorly, or not previously, described roles in the TCR cascade were identified, implicating PEP in regulating a broader range of signals than initially anticipated. While further work is required to fully characterize these substrates, our com-bined data indicate that expression of PEP-R619W leads to ampli-fication of proximal events within the TCR signaling cascade and also suggest that PEP-R619W expression versus PEP deficiency may differentially affect a subset of downstream effectors.
Finally, to investigate the regulation of antigen-specific T cell responses in vivo, we immunized mice with OVA (27). One week later, splenocytes were isolated and stimulated with OVA323–339
peptide and IL-2 production was quantified (Figure 3E). Cells derived from knockin mice produced more IL-2 than controls, demonstrating that PEP-R619W enhances antigen-specific T cell responses in vivo.
Altered B cell development in PEP-R619W knockin mice. Recent work from our group has shown that healthy individuals with the PTPN22 variant exhibit increased transitional B cell numbers and partial resistance of this population to B cell antigen recep-tor–induced (BCR-induced) apoptosis (9). To address whether PEP-R619W affects murine B cell homeostasis, we performed FACS analyses of BM and splenic B cell subsets. BM B cell devel-opment was unaltered in young mice (2 to 3 months; data not shown). However, the proportion and number of mature recircu-lating B cells (IgM+B220hi or B220+IgM+IgDhi) were decreased in
aged T/C and T/T mice (6 months; Figure 5, A and F). The reduc-tion in mature BM B cells did not correlate with any impairment in CD22 expression (Supplemental Figure 11C and refs. 28, 29). In the spleen, T/C or T/T mice displayed an increased percent-age and absolute numbers of transitional 1 (T1) and a slightly decreased proportion of follicular mature (FM) B cells at all ages (6 months, Figure 5, B and G; 2 to 3 months, Figure 6A). Thus, PEP-R619W leads to an increase in splenic T1 B cells and a mod-est decline in BM mature B cells.
B1 B cells are capable of generating pathogenic autoantibodies, and accumulation of this population has been reported in several autoimmune mouse models (30). To assess whether PEP-R619W altered B1 B cell development, we analyzed splenic and peritoneal cavity B1 B cells. T/T and WT mice had similar percentages of splenic B1 cells (CD5loIgM+) and peritoneal B1a cells (CD19+CD5lo
CD43+ or CD11b+IgM+CD5lo) and B1b cells (CD19+CD5–CD43+ or
CD11b+IgM+CD5–) (Supplemental Figure 13, A–C, and data not
shown) at all ages. T/T and WT mice also had comparable frequen-cies of peripheral blood B cells (Supplemental Figure 13D).
While aged PEP-R619W knockin and PEP-knockout mice share some features of dysregulated immune function, including enlarged spleens, expansion of memory/effector T cells and spon-taneous GC development, these strains also differ in important ways. First, both naive and memory/effector CD4+ T cells from
PEP-R619W mice were hyperresponsive to antigen stimulation, while only memory/effector T cells showed enhanced cell prolif-eration in PEP-deficient mice. Second, CpG prestimulated B cells from PEP-R619W mice exhibited modest alterations in proxi-mal BCR signaling not observed in PEP-deficient cells. Third, PEP-R619W mice, unlike PEP-deficient animals, displayed an expanded transitional B cell compartment that was more resistant, compared with WT control cells, to apoptosis in vivo and in vitro (data not shown). In addition, PEP-R619W mice in an “at risk” genetic background developed a wide range of autoantibodies and histological lesions not present in C57BL/6J PEP-deficient mice.
Although the reasons for these differences remain to be pre-cisely defined, one plausible explanation would be the capacity of PEP-R619W to access substrates not normally modified by the WT protein and inaccessible in the absence of PEP. In partial support of this idea, phosphoproteome analysis suggested that PEP-R619W expression led to alterations in proximal TCR substrates and other signaling effectors that differed markedly from WT and also subtly from PEP-deficient cells. Consistent with this theme, previous work indicates that the variant affects the capacity to interact with Csk (5) and additional work suggests that the signaling phenotype of LYP-R620W overexpressing cells may be altered by Csk coexpression (10). Intriguingly, our data suggest that PEP-R619W may affect the phosphorylation status of several SH3-containing proteins poten-tially capable of interacting with other proline-rich regions of PEP, including SKAP2, a previously identified substrate for LYP and oth-ers (26). The dramatic impact of R-to-W variant in both murine and human tolerance suggests that such subtle signaling differences in WT versus variant- or phosphatase-deficient cells are likely most relevant in physiological settings where response to low-abundance antigen and/or costimulatory signals is rate limiting.
In addition to previous work showing altered T cell function (8, 40), accumulating evidence indicates that LYP-R620W affects human B cell homeostasis (9, 41). Specifically, recent work from our laborato-ries shows that healthy individuals with the risk variant manifest an expansion of transitional and anergic B cells and that B cells from these subjects exhibit reduced apoptosis upon BCR engagement (9). Further, the risk allele affects central and/or peripheral human B cell tolerance, leading to accumulation of newly formed autoreac-tive cells (41). In line with these findings, PEP-R619W mice exhibit altered B cell homeostasis with expanded transitional, GC, and ABC B cells. PEP-R619W also alters B cell repertoire selection and results in enrichment of self-reactive PC-binding B cells. Most notably, B lin-eage–restricted expression of PEP-R619W was sufficient to trigger GC B cell activation, accumulation of ABC B cells, and some autoimmune features. Together, these data suggest that PEP-R619W likely disturbs B cell selection at the transitional and/or GC stages. Detailed reper-toire studies in the future should provide a precise characterization of the steps wherein PEP-R619W affects B cell selection.
The effect of LYP-R620W on AR-induced responses in human lymphoid cells remains controversial, with published data describ-ing both attenuated and enhanced signaldescrib-ing (8, 11). The current work best supports a model where PEP-R619W manifests as a dys-regulated variant that augments murine AR signaling in a manner partially distinct from PEP deficiency. We hypothesize that the par-features of autoimmunity, we generated Rosa HA-PEP-R619W
knockin mice wherein a cDNA encoding PEP-R619W and cis- linked GFP (using a 2A-linked self-cleaving linker) was introduced into the endogenous Rosa-26 locus (ref. 37 and Figure 7A). After breeding this strain with CD19-Cre mice, excision of the STOP-cassette efficiently induced HA-tagged PEP-R619W and GFP coexpression specifically within the B cell compartment (with PEP-R619W expressed at approximately one-half the level of the endogenous protein; Figure 7B).
Strikingly, while analysis of young Rosa PEP-R619W/CD19-Cre animals revealed no abnormalities, aged mice (10 months), unlike controls, developed splenomegaly and increased spleen cellular-ity (Figure 7C). Renal histopathology revealed glomerular lesions, including increased cellularity and mesangial matrix and infil-tration of Mac-2+ cells (Figure 7D). Rosa PEP-R619W/CD19-Cre
mice also developed anti-dsDNA autoantibodies (Figure 7E), and autoantigen array analysis revealed the presence of range of addi-tional autoantibodies similar to aged knockin animals (Supple-mental Figure 5B). While T cell development was not altered, Rosa PEP-R619W/CD19-Cre animals exhibited increases in GC and ABC B cells compared with controls (Figure 7, F and G, and data not shown). Together, these findings indicate PEP-R619W expres-sion in B cells is sufficient to initiate altered immune tolerance.
Discussion
The C1858T polymorphism in human PTPN22 is strongly asso-ciated with autoimmune diseases characterized by the presence of high-titer pathogenic autoantibodies (3, 4). Here, we generated mice with a knockin mutation in Ptpn22 designed to correspond to the human risk allele. In contrast to recent data showing dimin-ished PEP/LYP expression in cells expressing the relevant variant proteins (11), our data strongly support the conclusion that an R-to-W change within the proline-rich region 1 of this phosphatase does not impair protein expression, half-life, or calpain 1–medi-ated proteolysis. Aged PEP-R619W mice in a C57BL/6J and 129/Sv mixed genetic background developed autoimmune features char-acterized by expansion of activated and autoimmune prone B cells, increased numbers of effector/memory T cells, a broad array of high-titer autoantibodies, and vasculitic lesions in the lung and other organs. PEP-R619W expression augmented AR-dependent signaling and altered both T and B cell homeostasis. Perhaps most surprising, as based on previous speculation with respect to the key function of PEP/LYP in T cells, B lineage–restricted expression of PEP-R619W was sufficient to trigger autoimmunity.
(for T cells). Cells were then washed and stimulated with 10 μg/ml anti-IgM (for B cells), 5 μg/ml biotin-conjugated anti-CD3, and cross-linked by 5 μg/ml streptavidin (for splenic T cells) or 2 μg/ml biotin-conjugated anti-CD3 and cross-linked by 2 μg/ml streptavidin (for thymocytes). Induction of Ca2+ mobilization was determined by flow cytometry.
T and B cell proliferation. Splenic CD4+ T cells from 2- to 3-month-old
mice were purified by CD4+ T Cell Isolation Kit II (Miltenyi Biotec).
The enriched CD4+ T cells were stained with anti-CD4, anti-CD25,
anti-CD62L, and naive CD4+ (CD4+CD25–CD62Lhi) T cells, or effector/
memory CD4+ (CD4+CD25–CD62Llo/–) cells were sorted by FACSAria
(BD Biosciences). Sorted cells were labeled with CellTrace (Life Technol-ogies) for 20 minutes at 37°C and stimulated for 2–3 days with plate-bound anti-CD3 plus anti-CD28. To obtain in vitro–generated T cells, purified CD4+ T cells were stimulated with anti-CD3/CD28 for 3 days
followed by culture in IL-2–containing medium for an additional 2 days (13). Cells were labeled with CFSE as described (33) and stimulated with plate-bound anti-CD3. For B cell proliferation assays, splenic B cells were purified by the negative selection with anti-CD43 magnetic beads (Miltenyi Biotech). Purified B cells were labeled with CellTrace and stim-ulated for 3 days with 10 μg/ml anti-IgM (Jackson ImmunoResearch) with or without 1 μg/ml anti-CD40 (Southern Biotech), 1 μg/ml LPS (Sigma-Aldrich), or 100 nM CpG (InvivoGen). T and B cell proliferation were determined by CellTrace/CFSE dilution.
Western blotting. Western analyses of primary lymphocytes and cultured cells were performed as described previously (9, 42). The following antibod-ies were used in the study: anti-PEP P1 (against aa 588–654) and P2 (against aa 670–740) (gifts from A. Chan; Genentech); anti-LYP (against aa 306–684, MAB3428; R&D systems); anti-LYP (against aa 1–180, 4F6; Novus); anti– phospho-PLCγ2 (as described in ref. 42); anti-HA (6E2), anti–phospho-JNK (G9), anti–phospho-ERK (9101), anti–phospho-PLCγ1 (Y783), anti–phos-pho-Syk (C87C1), anti–phospho-IκBα (5A5), and anti-ERK (L34F12) from Cell Signaling Technology; anti-mouse β-actin (A-2066) and anti-FLAG (M2) from Sigma-Aldrich, and anti-pY (4G10) from Millipore.
ELISA and immunizations. Anti-dsDNA and anti-insulin–specific IgG ELISAs were performed as described previously (43, 44). Total ANA Ig in supernatants of cultured thymic B cells was determined by the ANA ELISA kit (5210) from Alpha Diagnostic International Inc. A broad range of IgM and IgG autoantibodies was assessed by autoantigen arrays (UT South-western Medical Center, Microarray Core Facility, Dallas, Texas, USA) as described (45). The complete autoantigen array data sets are provided in Supplemental Figure 5. Total serum levels of IgM, IgG, IgG1, IgG2a, IgG2b, IgG3, and IgA were determined by ELISA kit from Southern Bio-tech. IgE levels were determined by IgE ELISA kit from BD Biosciences. Serum TNP- or NP-specific antibodies were measured by ELISA with plate-bound TNP-BSA or NP-BSA (Biosearch Tech) and then detected by iso-type-specific HRP-conjugated secondary antibodies (Southern Biotech).
Statistics. Statistical significance of comparisons of mean values was assessed by a 2-tailed Student’s t test or nonparametric Mann-Whitney U
test and, in the case of multiple experimental group comparisons, by 1-way ANOVA with Bonferroni’s correction using GraphPad software. P < 0.05 was considered significant.
Study approval. All animal protocols were approved by the Institutional Animal Care and Use Committee of Seattle Children’s Research Institute.
Additional methods including generation of knockin animals, flow cytometry, Treg suppression assays, immunization, and detailed phospho- proteomic analysis are provided in Supplemental Methods.
Acknowledgments
We thank K. Sommer and K. Hudkins for technical assistance; T. Jahan for sharing the Tagtrac program for analyzing phospho- adoxically “attenuated” signals described in primary human
mem-ory T and B cells reflects the longer-term outcome of enhanced signaling that ultimately leads to hyporesponsiveness. Consistent with this idea, memory/effector T cells (CD44hiCD62Llo/–) enriched
in aged mice were minimally responsive to TCR-induced calcium flux (data not shown). Future studies using risk-variant naive T and B cells derived from human cord blood cells and/or thymus may be the most reliable way to clarify this issue.
In summary, our data indicate that PEP-R619W does not alter protein stability, but dysregulates its activity, with the consequence of enhanced AR signaling. Our findings strongly suggest that these modified signals directly affect the naive T and B cell repertoire via altered negative and/or positive selection. As shown here, PEP expres-sion is markedly upregulated within activated B and T cells, imply-ing that dysregulated PEP signals may also affect GC responses and thereby function to iteratively modulate T and B cell selection and break tolerance in an “at risk” genetic background. Further mecha-nistic analysis of how altered PEP/LYP localization modulates lym-phoid signals may identify novel therapeutic targets for treatment of the autoimmune diseases associated with this common risk allele.
Methods
Mice. The generation of PEP-R619W knockin mice and Rosa PEP-R619W knockin mice is described in detail in Supplemental Methods. OT-II mice (B6.Cg-Tg [TcraTcrb] 425Cbn/J) were from Jackson Laboratory. M167 heavy-chain transgenic mice (line U243-4) were provided by S. Porcelli (Albert Einstein College of Medicine, New York, New York, USA). PEP-defi-cient mice and HY-TCR transgenic mice were provided by A. Chan (Genen-tech). PEP-R619W knockin mice were back-crossed to C57BL/6J for 5 gen-erations before crossing to M167, OT-II, or HY-TCR transgenic mice. The experimental mice contained 1 copy of M167, OT-II, or HY-TCR transgene, respectively. Mice were maintained in a specific pathogen–free facility.
In vitro CHX treatment. N-terminal HA-tagged or FLAG-tagged PEP and PEP-R619W or LYP and LYP-R620W (provided by A. Weiss, UCSF, San Francisco, California, USA) were overexpressed in Ramos B cells or Jur-kat T cells by lentiviral-mediated gene transfer (9). Cells were treated with 50 μM CHX for 0–30 minutes at 37°C, and the total cell lysates were sub-jected to Western blot analysis.
In vitro calpain 1 cleavage assay. Cleavage assays were performed as described previously, with minor modifications (11). Anti-FLAG M2 affin-ity gel (F2426; Sigma-Aldrich) was used to IP FLAG-tagged proteins from Ramos B cells or Jurkat T cells overexpressing FLAG-tagged PEP or LYP. IPs were incubated for 0–30 minutes at 37°C with 0.05 U calpain 1 (EMD Biosciences) in activation buffer (40 mM HEPES, 10 mM DTT, 0.5 mM CaCl2). The reactions were boiled in ×2 SDS-PAGE loading buffer, and the
supernatants were loaded on SDS-PAGE gels for Western blot analysis.
Histology analysis. Tissues from 6-month-old PEP-R619W knockin mice and WT littermates were fixed in 10% neutral buffered formalin, embed-ded in paraffin, sectioned, and stained with H&E. Sections were examined by a board-certified veterinary pathologist who was blinded to the study design. Photomicrographs were taken using a Nikon DS-Fi1 digital cam-era using NIS-elements software.
STZ-induced diabetes. Six- to eight-week-old male mice were injected intraperitoneally with 40 mg/kg of STZ (Sigma-Aldrich) on 4 consec-utive days (18). Blood glucose levels were measured twice weekly using Bayer Contour Meter (Bayer) beginning at 2 weeks after the final STZ injection. Diabetes was defined by the elevation of glucose levels above 250 mg/dl for 2 consecutive tests.
Received for publication September 20, 2012, and accepted in revised form February 7, 2013.
Address correspondence to: David J. Rawlings, Center for Immunity and Immunotherapies, Seattle Children’s Research Institute, 1900 Ninth Avenue, Seattle, Washington 98101, USA. Phone: 206.987.7319; Fax: 206.987.7310; E-mail: drawling@u.washington.edu.
proteomic data; and members of the Rawlings laboratory for valu-able suggestions and discussions. This work was supported by a JDRF Center grant (no. 4-2007-1058); NIH grants HD037091, HL075453, AI084457, AI071163 (to D.J. Rawlings), and K99-HL103768 (to. R.G. James); and the University of Washington’s Proteomics Resource (UWPR95794). R.T. Moon is an investigator of the Howard Hughes Medical Institute.
1. Bottini N, et al. A functional variant of lymphoid tyrosine phosphatase is associated with type I dia-betes. Nat Genet. 2004;36(4):337–338.
2. Bottini N, Vang T, Cucca F, Mustelin T. Role of PTPN22 in type 1 diabetes and other autoimmune diseases. Semin Immunol. 2006;18(4):207–213. 3. Vang T, Miletic AV, Arimura Y, Tautz L, Rickert RC,
Mustelin T. Protein tyrosine phosphatases in auto-immunity. Annu Rev Immunol. 2008;26:29–55. 4. Chung SA, Criswell LA. PTPN22: its role in SLE and
autoimmunity. Autoimmunity. 2007;40(8):582–590. 5. Vang T, et al. Autoimmune-associated lymphoid
tyrosine phosphatase is a gain-of-function variant.
Nat Genet. 2005;37(12):1317–1319.
6. Cloutier JF, Veillette A. Cooperative inhibition of T-cell antigen receptor signaling by a complex between a kinase and a phosphatase. J Exp Med. 1999; 189(1):111–121.
7. Cohen S, Dadi H, Shaoul E, Sharfe N, Roifman CM. Cloning and characterization of a lym-phoid-specific, inducible human protein tyrosine phosphatase, Lyp. Blood. 1999;93(6):2013–2024. 8. Rieck M, Arechiga A, Onengut-Gumuscu S,
Green-baum C, Concannon P, Buckner JH. Genetic variation in PTPN22 corresponds to altered function of T and B lymphocytes. J Immunol. 2007;179(7):4704–4710. 9. Habib T, et al. Altered B cell homeostasis is
associ-ated with type I diabetes and carriers of the PTPN22 allelic variant. J Immunol. 2012;188(1):487–496. 10. Zikherman J, Hermiston M, Steiner D, Hasegawa
K, Chan A, Weiss A. PTPN22 deficiency cooperates with the CD45 E613R allele to break tolerance on a non-autoimmune background. J Immunol. 2009; 182(7):4093–4106.
11. Zhang J, et al. The autoimmune disease-associated PTPN22 variant promotes calpain-mediated Lyp/ Pep degradation associated with lymphocyte and dendritic cell hyperresponsiveness. Nat Genet. 2011; 43(9):902–907.
12. Arechiga AF, et al. Cutting edge: the PTPN22 allelic variant associated with autoimmunity impairs B cell signaling. J Immunol. 2009;182(6):3343–3347. 13. Hasegawa K, Martin F, Huang G, Tumas D, Diehl
L, Chan AC. PEST domain-enriched tyrosine phosphatase (PEP) regulation of effector/memory T cells. Science. 2004;303(5658):685–689. 14. Scadding GK, Vincent A, Newsom-Davis J, Henry
K. Acetylcholine receptor antibody synthesis by thymic lymphocytes: correlation with thymic his-tology. Neurology. 1981;31(8):935–943.
15. O’Reilly LA, et al. Studies on the thymus of non -obese diabetic (NOD) mice: effect of transgene expression. Immunology. 1994;82(2):275–286. 16. Miyama-Inaba M, et al. Unusual phenotype of B
cells in the thymus of normal mice. J Exp Med. 1988; 168(2):811–816.
17. Sato T, et al. Aberrant B1 cell migration into the thymus results in activation of CD4 T cells through its potent antigen-presenting activity in the devel-opment of murine lupus. Eur J Immunol. 2004; 34(12):3346–3358.
18. Lamhamedi-Cherradi SE, Zheng SJ, Maguschak KA, Peschon J, Chen YH. Defective thymocyte apoptosis and accelerated autoimmune diseases in TRAIL-/- mice. Nat Immunol. 2003;4(3):255–260. 19. Zikherman J, et al. CD45-Csk phosphatase-kinase
titration uncouples basal and inducible T cell receptor signaling during thymic development.
Immunity. 2010;32(3):342–354.
20. Barnden MJ, Allison J, Heath WR, Carbone FR. Defective TCR expression in transgenic mice con-structed using cDNA-based alpha- and beta-chain genes under the control of heterologous regulatory elements. Immunol Cell Biol. 1998;76(1):34–40. 21. Lu LF, Rudensky A. Molecular orchestration of
differentiation and function of regulatory T cells.
Genes Dev. 2009;23(11):1270–1282.
22. Maine CJ, et al. PTPN22 alters the development of regulatory T cells in the thymus. J Immunol. 2012; 188(11):5267–5275.
23. Fu G, et al. Phospholipase Cγ1 is essential for T cell development, activation, and tolerance. J Exp Med. 2010;207(2):309–318.
24. Zhang Y, et al. Time-resolved mass spectrometry of tyrosine phosphorylation sites in the epider-mal growth factor receptor signaling network reveals dynamic modules. Mol Cell Proteomics. 2005; 4(9):1240–1250.
25. Wu J, et al. Identification of substrates of human protein-tyrosine phosphatase PTPN22. J Biol Chem. 2006;281(16):11002–11010.
26. Yu X, et al. Substrate specificity of lymphoid-spe-cific tyrosine phosphatase (Lyp) and identifica-tion of Src kinase-associated protein of 55 kDa homolog (SKAP-HOM) as a Lyp substrate. J Biol Chem. 2011;286(35):30526–30534.
27. Zhang Y, et al. Regulation of T cell activation and tolerance by PDL2. Proc Natl Acad Sci U S A. 2006; 103(31):11695–11700.
28. Nitschke L, Floyd H, Ferguson DJ, Crocker PR. Iden-tification of CD22 ligands on bone marrow sinus-oidal endothelium implicated in CD22-dependent homing of recirculating B cells. J Exp Med. 1999; 189(9):1513–1518.
29. Tedder TF, Poe JC, Haas KM. CD22: a multifunc-tional receptor that regulates B lymphocyte sur-vival and signal transduction. Adv Immunol. 2005; 88:1–50.
30. Hayakawa K, Hardy RR. Development and function of B-1 cells. Curr Opin Immunol. 2000;12(3):346–353. 31. Kenny JJ, O’Connell C, Sieckmann DG, Fischer
RT, Longo DL. Selection of antigen-specific, idio-type-positive B cells in transgenic mice expressing a rearranged M167-mu heavy chain gene. J Exp Med. 1991;174(5):1189–1201.
32. Kenny JJ, et al. Receptor-mediated elimination of phosphocholine-specific B cells in x-linked immune-deficient mice. J Immunol. 1991; 146(8):2568–2577.
33. Meyer-Bahlburg A, Andrews SF, Yu KO, Porcelli SA, Rawlings DJ. Characterization of a late transitional B cell population highly sensitive to BAFF-medi-ated homeostatic proliferation. J Exp Med. 2008; 205(1):155–168.
34. Vinuesa CG, Sanz I, Cook MC. Dysregulation of germinal centres in autoimmune disease. Nat Rev Immunol. 2009;9(12):845–857.
35. Rubtsov AV, et al. Toll-like receptor 7 (TLR7)-driven accumulation of a novel CD11c(+) B-cell population is important for the development of autoimmunity. Blood. 2011;118(5):1305–1315. 36. Wen R, et al. Phospholipase Cgamma2
pro-vides survival signals via Bcl2 and A1 in differ-ent subpopulations of B cells. J Biol Chem. 2003; 278(44):43654–43662.
37. Srinivas S, et al. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol. 2001;1:4.
38. Behrens TW. Lyp breakdown and autoimmunity.
Nat Genet. 2011;43(9):821–822.
39. Vang T, et al. LYP inhibits T-cell activation when dis-sociated from CSK. Nat Chem Biol. 2012;8(5):437–446. 40. Rhee I, Veillette A. Protein tyrosine phosphatases in lymphocyte activation and autoimmunity. Nat Immunol. 2012;13(5):439–447.
41. Menard L, et al. The PTPN22 allele encoding an R620W variant interferes with the removal of devel-oping autoreactive B cells in humans. J Clin Invest. 2011;121(9):3635–3644.
42. Andrews SF, Rawlings DJ. Transitional B cells exhibit a B cell receptor-specific nuclear defect in gene tran-scription. J Immunol. 2009;182(5):2868–2878. 43. Becker-Herman S, et al. WASp-deficient B cells play
a critical, cell-intrinsic role in triggering autoim-munity. J Exp Med. 2011;208(10):2033–2042. 44. Michel C, Boitard C, Bach JF. Insulin
autoanti-bodies in non-obese diabetic (NOD) mice. Clin Exp Immunol. 1989;75(3):457–460.