0095-1137/96/$04.0010
Copyrightq1996, American Society for Microbiology
Comparison of Ribotyping, Arbitrarily Primed PCR, and
Pulsed-Field Gel Electrophoresis for Molecular Typing
of Listeria monocytogenes
M. LOUIE,
1* P. JAYARATNE,
2I. LUCHSINGER,
2J. DEVENISH,
1J. YAO,
1W. SCHLECH,
3AND
A. SIMOR
1Department of Microbiology, SD Laboratory Services, Sunnybrook Health Science Centre, University of Toronto,
Toronto, Ontario,
1Hamilton General Hospital, Hamilton, Ontario,
2and Victoria General
Hospital, Halifax, Nova Scotia,
3Canada
Received 22 June 1995/Returned for modification 3 August 1995/Accepted 5 October 1995
Fifty-one clinical isolates of
Listeria monocytogenes
(15 isolates from two outbreaks and 36 epidemiologically
unrelated isolates) were typed by conventional serotyping, ribotyping (RT), pulsed-field gel electrophoresis
(PFGE), and arbitrarily primed PCR (AP-PCR). Serotyping was unable to distinguish between related and
unrelated strains of
L. monocytogenes
. Each of the three molecular methods showed excellent typeability and
reproducibility. Restriction with
Eco
RI and
Pvu
II gave 16 and 23 RT patterns, respectively. Restriction with
Apa
I or
Sma
I generated 22 and 26 PFGE profiles, respectively.
Apa
I profiles were easier to interpret, with 10
to 15 bands each, while
Sma
I profiles had 15 to 20 bands each. AP-PCR with two different primers yielded 29
and 31 randomly amplified polymorphic DNA patterns, respectively. Strains from the same outbreak shared
concordant patterns by each of the three methods. Of the three techniques evaluated, RT was the least
discriminating and could not distinguish between strains from the two outbreaks. The abilities of AP-PCR and
PFGE to differentiate between strains were comparable. However, AP-PCR was more rapid and easier to
perform. We conclude that the DNA profiles generated by either AP-PCR or PFGE can be used to differentiate
outbreak strains from epidemiologically unrelated strains and to clearly identify unrelated strains as being
distinct from one another. We recommend that at least two independent primers be used for AP-PCR typing
in order to improve its discriminatory power.
Listeria monocytogenes is recognized as an important human
pathogen causing food-borne outbreaks and sporadic
infec-tions. It may cause invasive disease such as bacteremia,
men-ingitis, and severe perinatal infections (12). Several large
food-borne outbreaks have incriminated commercial food products
as a primary source of infection for both epidemic and sporadic
listerioses (25). Because of the ubiquitous nature of L.
mono-cytogenes in the environment and its potential presence in
multiple food sources, highly discriminatory typing systems are
necessary for epidemiological investigations.
Serotyping of L. monocytogenes strains is not very
discrimi-natory since almost all strains isolated from humans, foods,
and environments belong to a small number of serotypes (11,
12, 22, 25). Other techniques used for typing L. monocytogenes
have included biotyping, phage typing, and multilocus enzyme
electrophoresis. None of these methods are capable of typing
and differentiating all strains, nor are they readily available in
nonreference centers (1, 5). More recently, genotypic
tech-niques such as plasmid profiling, ribotyping (RT), analysis of
chromosomal DNA by either restriction enzyme analysis or
pulsed-field gel electrophoresis (PFGE), and fingerprinting by
arbitrarily primed PCR (AP-PCR) have been used for typing
purposes. These techniques are much more applicable for use
in the routine clinical laboratory. However, few reports of
studies comparing the application of these various molecular
typing methods for L. monocytogenes have been published (8,
13, 21, 22).
In the study described here we compared RT, PFGE, and
AP-PCR for their abilities to differentiate between
epidemio-logically related and unrelated serotypes of L. monocytogenes.
We also evaluated these methods for their reproducibilities
and their ease of use in the clinical laboratory.
(This study was presented in part at the 34th Interscience
Conference on Antimicrobial Agents and Chemotherapy,
Or-lando, Fla., 4 to 7 October 1994 [15a].)
MATERIALS AND METHODS
Bacterial strains.L. monocytogenes strains were randomly selected from two well-documented outbreaks from Nova Scotia (12 isolates) (24) and California (3 isolates) (15). Isolates of epidemiologically unrelated L. monocytogenes strains (36 isolates) were collected between 1974 and 1993 from the blood and cerebro-spinal fluid of patients at three different geographic centers in Ontario (Toronto, Hamilton, and Ottawa).
The strains were identified as L. monocytogenes by typical Gram stain appear-ance, colonial morphology, bile esculin hydrolysis, and characteristic tumbling motility (6). The isolates were stored at2708C in buffered glycerol and were subcultured onto blood agar twice prior to testing.
Serotyping.Strains from the Nova Scotia and California outbreaks were pre-viously serotyped by the Centers for Disease Control and Prevention, Atlanta, Ga. Epidemiologically unrelated L. monocytogenes strains from Ontario were serotyped at the Special Bacteriology Section, Laboratory Centre for Disease Control, Ottawa, Ontario, Canada, by using the procedures outlined by Seeliger and Hohne (26).
RT.Genomic DNA was extracted by the method of Ausubel et al. (2), and 2 to 3mg of purified DNA from each study isolate was ribotyped with two restric-tion endonucleases (EcoRI and PvuII) according to the manufacturer’s (Pro-mega Co., Madison, Wis.) recommendations. Following digestion, the DNA fragments were separated by agarose gel electrophoresis and were transferred to nylon membranes (Boehringer Mannheim, Laval, Quebec, Canada) by the method of Southern blotting (23). The transferred DNA was fixed to the mem-brane by UV cross-linking (UV Crosslinker FB-UVXL-1000; Fisher Biotech, Ottawa, Ontario, Canada).
Plasmid pKK3535, containing the rrnB ribosomal operon of Escherichia coli (9), was used as the DNA probe for ribotyping. Linearized plasmid DNA was * Corresponding author. Mailing address: Department of
Microbi-ology, B-121, Sunnybrook Health Science Centre, 2075 Bayview Ave., Toronto, Ontario M4N 3M5. Canada. Phone: (416) 480-4242. Fax: (416) 480-6845.
15
on May 15, 2020 by guest
http://jcm.asm.org/
labelled with digoxigenin (DIG)-11-dUTP by using the DIG DNA Labeling Kit (Boehringer Mannheim). Southern hybridizations were performed under condi-tions of low stringency at 378C overnight in the presence of 30% formamide (Bethesda Research Laboratories Inc., Gaithersburg, Md.). The membranes were washed twice for 5 min each time at room temperature with 23SSC (13 SSC is 0.15 M NaCl plus 0.015 M sodium citrate [pH 7.0])–0.1% sodium dodecyl sulfate (SDS; Bethesda Research Laboratories) and twice for 15 min each time at 378C with 0.13SSC–0.1% SDS. Hybridization signals were detected with the DIG DNA Labeling and Detection Kit (Boehringer Mannheim) according to the manufacturer’s recommendations.
Ribotypes were considered identical only if they exhibited the same number and size of all bands in their profiles.
PFGE.Each strain of L. monocytogenes was grown overnight in 5 ml of brain heart infusion broth. After centrifugation, each cell pellet was suspended in 750 ml of 10 mM Tris-HCl (pH 7.6)–1 M NaCl buffer. Agarose plugs were made from a 1:1 mixture of 1.6% low-melting-point agarose (Gibco-BRL, Burlington, On-tario, Canada) and the cell suspension. Each plug was lysed in lysis buffer (6 mM Tris-HCl [pH 7.6; Sigma Chemical Co., St. Louis, Mo.], 1 M NaCl [Sigma], 100 mM EDTA [pH 9; Sigma], 0.5% Brij-58 [Sigma], 0.2% deoxycholic acid [Sigma], 0.5% Sarkosyl [Sigma], 20mg of RNase [Sigma] per ml, and 1 mg of lysozyme [Sigma] per ml) for 18 h at 378C. The samples were then treated for 16 h at 508C with the same volume of solution containing 100mg of proteinase K (Boehringer Mannheim) per ml, 0.5% Sarkosyl, and 0.5 M EDTA (pH 9). After three 1-h washes with TE buffer (10 mM Tris-HCl [pH 7.6], 0.1 mM EDTA [pH 9]), the agarose plugs were incubated for 20 h with one of the restriction endonuclease enzymes ApaI or SmaI (Boehringer Mannheim) according to the manufacturer’s recommendations. All study isolates were typed by both endonucleases. The resultant DNA fragments were electrophoresed on a 1% PFGE agarose (Bio-Rad Laboratories, Mississauga, Ontario, Canada) gel in a contour-clamped ho-mogeneous electrical field by using the CHEF DR-II system (Bio-Rad, Hercules, Calif.), with 0.53TBE buffer at 6 V/cm and 128C. With ApaI restriction, the pulse times were linearly ramped from 1 to 35 s over 22 h; with SmaI digestion, the pulse times ranged from 0.2 to 25 s over 20 h with linear ramping.
Strains with one or two band shifts consistent with a single genetic event were considered to be clonally related and subtypes of each other (17). Strains that differed by three or more bands were considered to represent unique strains.
AP-PCR.The random oligonucleotide primers PJ108 (59-GCTTATTCTTGA CATCCA-39) and PJ118 (59-TGTTCGTGCTGTTTCTG-39) (Vetrogen Co., London, Ontario, Canada) were selected from a group of 10 primers on the basis of their performance in trial experiments to produce reproducible, randomly amplified polymorphic DNA (RAPD) patterns (unpublished data) and were used for subsequent AP-PCR amplifications. Template DNA was prepared and extracted by the method described by Ausubel et al. (2). Amplification reaction mixtures contained 25 ng of genomic DNA, 200 ng of the respective oligonucle-otide primer, 200mM deoxynucleoside triphosphates, 2.5 mM MgCl2, and 2.5 U of Taq polymerase (Promega Co.) in a 50-ml volume. Amplification was
per-formed with a Perkin-Elmer DNA Thermal Cycler 480 (Perkin-Elmer Cetus, Norwalk, Conn.) with temperature ramping as follows: two cycles at 948C for 5 min, 408C for 5 min, and 728C for 5 min and then 40 cycles of 948C for 1 min, 408C for 2 min, and 728C for 2 min. In the final cycle, the polymerization step was extended to 10 min. Negative controls were prepared and amplified similarly, but without the addition of target DNA. Randomly amplified products (15ml) were size separated on 1.2% agarose gels by electrophoresis and were visualized with ethidium bromide.
The RAPD patterns generated by AP-PCR were considered identical on the basis of similar numbers and matching positions of all bands. A single band difference was sufficient to differentiate between strains and to assign a specific RAPD profile.
Reproducibility and discriminatory power of the three typing mehtods.L. monocytogenes strains were tested on at least two separate occasions by all three molecular typing methods to assess the reproducibilities of the typing methods. The unique banding profiles generated by each of the typing methods were arbitrarily given serial alphabetical or numerical designations. For PFGE, strains considered to be subtypes of each other were labelled with the same alphabetical designation followed by a different numerical designation for each subtype. All electrophoretic mobility gels included molecular weight size standards to facili-tate comparisons. As well, subtype categorization was confirmed by running the isolates with similar banding patterns on the same gel. Determination of banding profiles was made visually.
The discriminatory power of each typing method was determined by calculat-ing the discriminatory index (DI) by the method of Hunter and Gaston (14).
RESULTS
A total of 51 strains of L. monocytogenes including 15
epi-demiologically related strains from two separate outbreaks and
36 unrelated clinical strains from different geographic locations
were examined. The epidemiologically related strains were
previously typed as serotype 4b. The serotype distributions for
the unrelated L. monocytogenes strains included serotypes 4b
(n
5
14 isolates), 1/2a (n
5
12), 1/2b (n
5
7), 4c (n
5
2), and
4d (n
5
1).
[image:2.612.57.557.91.310.2]All L. monocytogenes strains were typeable by RT, PFGE,
and AP-PCR. The DNA typing profiles of the same isolate,
generated by each of the three molecular techniques, were
found to be stable and reproducible on at least two or more
separate occasions (data not shown). When discrepancies with
the interpretation of the DNA banding profiles occurred, the
TABLE 1. Typing patterns by serotyping, RT, PFGE, and AP-PCR of 15 epidemiologically related and 36 epidemiologically unrelated clinicalstrains of L. monocytogenes
Strain (no. of isolates) and
source
Serotype (no. of isolates)
RT (no. of isolates)a PFGE (no. of isolates)a,b AP-PCR (no. of isolates)a
EcoRI PvuII ApaI SmaI PJ108 PJ118
Related (15)
Nova Scotia 4b (11) I (11) I (11) A1 (7), A2 (3),
X (1)
A1 (7), A2 (3), X (1)
a (10), X (1) a (10), X (1)
1/2a (1) II (1) X (1) X (1) X (1) X (1) X (1)
California 4b (3) I (3) I (3) B (3) B (3) a (3) b (3)
Unrelated (36)
4b (14) I (4), II (1), III (3), IV (1), X (5)
II (5), III (4), X (5)
C1 (2), C2 (1), D1 (1), D2 (1), E1 (1), F1 (3), F2 (3), X (2)
C1 (2), C2 (1), D1 (3), D2 (1), D3 (1), X (6)
b (2), c (3), d (2), X (7)
c (2), d (1), e (2), f (1), X (8)
1/2a (12) V (5), VI (2), VII (2), X (3)
IV (4), V (2), VI (1), X (5)
G1 (3), G2 (3), G3 (1), X (5)
E1 (4), E2 (3), X (5)
e (3), f (2), X (7)
f (1), g (3), h (2), i (2), X (4) 1/2b (7) I (1), II (1),
VII (5)
VI (1), VII (3), VIII (2), X (1)
E2 (1), H (2), X (4)
F (2), X (5) g (2), X (5) j (2), X (5)
4c (2) I (1), X (1) X (2) C2 (1), X (1) X (2) b (1), X (1) X (2)
4d (1) IV (1) X (1) F2 (1) D1 (1) d (1) d (1)
a
X represents a distinct isolate with an unique DNA profile as generated by each of the molecular typing method. The number of single isolates with a distinct typing pattern is shown in parentheses.
b
Major PFGE DNA profiles are designated by a letter, and their clonal subtypes are designated by numerical suffixes.
on May 15, 2020 by guest
http://jcm.asm.org/
experiments were repeated and isolates with similar DNA
banding patterns were rerun on the same gel for direct
com-parison. Table 1 shows the typing patterns generated by the
three methods for the strains evaluated and their respective
serotypes. For each typing method, single isolates of L.
mono-cytogenes had distinct and unique profiles that were not shared
by other isolates (Table 1).
Among the 51 strains of L. monocytogenes, digestion with
EcoRI and PvuII gave 16 and 23 RT patterns, respectively. The
RT patterns generally had 8 to 15 bands ranging between 12
and 1 kb for EcoRI and between 20 and 2 kb for PvuII. RT
patterns were not serotype specific. Strains of serotypes 1/2b,
4b, and 4c shared EcoRI RT profile I; strains of serotypes 1/2a,
1/2b, and 4b shared EcoRI RT profile II; and one strain each
of serotypes 4b and 4d had an identical EcoRI profile, profile
IV. Strains of serotypes 1/2a and 1/2b also shared an identical
EcoRI profile, profile VII. Only one strain each of serotypes
1/2a and 1/2b shared an identical PvuII RT profile, profile VI,
while strains of serotypes 4b, 4c and 4d all had distinctly
dif-ferent PvuII RT profiles. For both enzymes, however, there
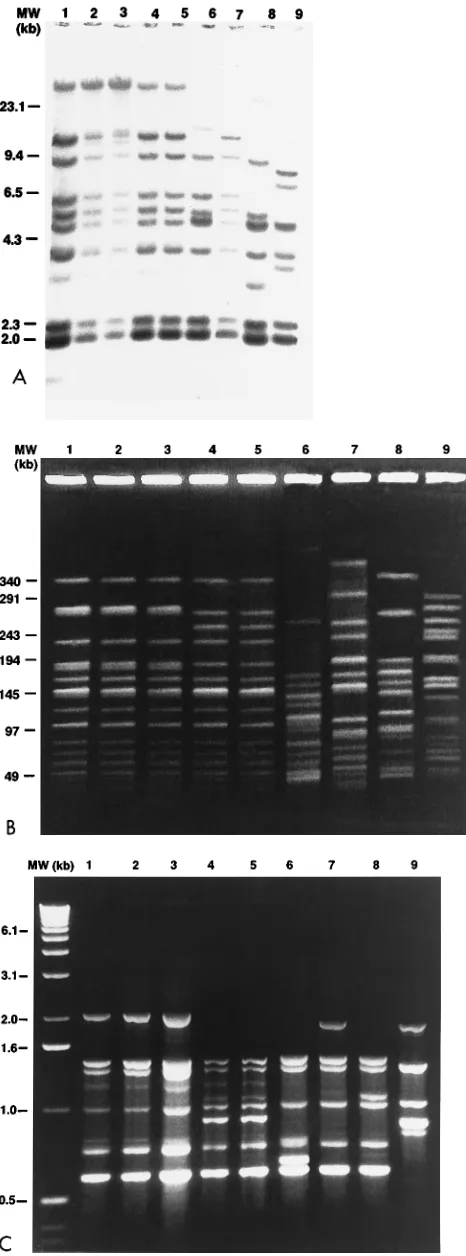
were several RT patterns within each serotype. Figure 1A
shows the RT patterns of EcoRI-restricted DNAs for both
related and unrelated L. monocytogenes strains. The presence
of an undigested chromosomal DNA band can be seen in lanes
1 to 5. This may be the result of excess DNA or a low level of
enzyme activity because of inhibitors present in the
chromo-somal DNA preparations.
PFGE generated 22 and 26 unique DNA fragment profiles
with ApaI and SmaI restriction endonuclease digestions,
re-spectively. The ApaI profiles were easier to interpret, with 10
to 15 bands each, while the SmaI profiles had 15 to 20 bands
each (data not shown). Of the 22 ApaI PFGE patterns, 16 were
unique. Each of five of the remaining six ApaI PFGE profiles
consisted of two subtypes, while in the other profile, three
subtypes were identified. There were 22 of 26 SmaI PFGE
profiles that were distinct. Of the remaining four SmaI PFGE
profiles, each of three profiles was further separated into two
subtypes. The other SmaI PFGE profile had three subtypes.
Strains of serotype 4b shared ApaI PFGE profiles (profiles C,
E, and F) with the profiles of isolates of serotypes 4c, 4d, and
1/2b. For SmaI PFGE profiles, only isolates of serotypes 4b
and 4d shared an identical PFGE profile (D1).
AP-PCR with two different primers, PJ108 and PJ118, gave
29 and 31 independent patterns, respectively. Both primers
yielded 6 to 10 amplified products ranging in size from 2.5 to
0.3 kb. With primer PJ108, strains of serotype 4b shared
iden-tical profiles (profiles b and d) with strains of serotypes 4c and
4d. Among the primer PJ118 profiles, serotype 4b had profiles
(profiles d and f) identical to those of serotypes 1/2a and 4d.
The reproducibility of the AP-PCR method was examined in
all isolates by three independent amplification reactions and
then agarose gel electrophoresis. No change in the DNA
fin-gerprints was observed in any of the three independent
exper-iments (data not shown). The use of boiled cell suspensions as
template DNA for amplification also produced results
compa-FIG. 1. DNA profiles of related and unrelated strains of L. monocytogenes. Results for the same nine L. monocytogenes strains are shown in each panel. Each panel includes molecular size markers (MW; in kilobases). Lanes 1 to 3,
representative Nova Scotia outbreak serotype 4b strains; lanes 4 and 5, California outbreak serotype 4b strains; lanes 6 and 7, epidemiologically unrelated serotype 4b strains; lane 8, serotype 1/2a strain; lane 9, serotype 1/2b strain. (A) RT patterns of EcoRI-restricted DNA. Lanes 1 to 5 and 7, profile I; lanes 6, 8, and 9, profiles X, V, and VII, respectively. Undigested chromosomal DNA can be seen in lanes 1 to 5. (B) PFGE profiles of ApaI-digested DNA. Lanes 1 to 3, profile A; lanes 4 and 5, profile B; lanes 6 to 9, unique profiles F1, D1, G1, and X, respectively. (C) AP-PCR profiles obtained with primer PJ118. Lanes 1 to 3, profile a; lanes 4 and 5, profile b; lanes 6 to 9, unique profiles f, d, g, and X, respectively.
on May 15, 2020 by guest
http://jcm.asm.org/
[image:3.612.61.294.71.699.2]rable to those obtained with purified genomic DNA (data not
shown). Negative control reactions performed with each
primer were consistently negative.
Comparison of typing methods.
Strains from the same
out-breaks (15, 24) shared concordant patterns by each of the three
molecular methods. Among the outbreak strains, all of which
were serotype 4b, PFGE by both ApaI and SmaI, and AP-PCR
with primer PJ118 were the most discriminating typing
meth-ods. RT was unable to differentiate between strains from the
two separate outbreaks. Two Nova Scotia strains, initially
thought to be a part of the original outbreak, were found on
PFGE and AP-PCR to have very different DNA profiles
com-pared with those of the other Nova Scotia outbreak strains. A
retrospective review of these two strains showed that they were
not a part of the original outbreak. The DI values for each of
the typing methods are given in Table 2.
There were several instances in which Listeria strains (12 of
36 strains) had DNA typing profiles that were concordant and
in agreement by all three methods, suggesting that they may
have been related. However, these strains were isolated from
clinically unrelated patients remote from one another both in
time and in place.
Representative DNA profiles of related and unrelated L.
monocytogenes strains by the three typing methods are shown
in Fig. 1.
DISCUSSION
Epidemiologic investigations of both endemic and epidemic
listerioses depend on the availability and reliability of highly
discriminatory typing systems which may differentiate between
strains and strains from different sources. The purpose of the
present study was to evaluate a set of epidemiologically related
and unrelated L. monocytogenes strains by three DNA-based
techniques (RT, PFGE, and AP-PCR) and to compare the
results with those obtained by serotyping.
Only a few serotypes, namely, serotypes 1/2a, 1/2b, and 4b,
have been involved in outbreaks and as causes of clinical
dis-ease (3, 20, 22). In the present study, these serotypes were also
the most commonly observed serotypes among the isolates
investigated. Specific differentiation of strains will require
con-firmation of the results obtained by complementary
tech-niques.
In the present study, there was a remarkable degree of
uniformity in the typing results obtained by RT, PFGE, and
AP-PCR, with excellent typeability and reproducibility. Of the
three molecular methods that were compared, RT was the
least discriminating for evaluating outbreak strains and
epide-miologically unrelated L. monocytogenes strains. As has been
previously reported, RT is less discriminating than restriction
enzyme analysis, multilocus enzyme electrophoresis, and
bac-teriophage typing (3, 13, 22). As a single typing system, RT is
not adequate for typing L. monocytogenes strains. The
addi-tional requirement of Southern blot analysis makes RT tedious
and time-consuming and requires facilities that may not be
readily available. Graves et al. (13) found that both RT and
multilocus enzyme electrophoresis did not provide adequate
discrimination between strains of different serotypes and
sug-gested that alternative methods such as PFGE and RAPD
analysis be considered.
PFGE provides an alternative tool for analyzing relatedness
among strains or species. Macrorestriction fingerprinting by
PFGE gives clear and reproducible resolution of fragments
that can be easily read by visual inspection. In the present
study, the PFGE profiles of ApaI macrorestriction of L.
mono-cytogenes DNA generated fewer bands and were easier to read
and interpret than SmaI-restricted DNA profiles. Both
en-zymes provided highly discriminatory patterns, allowing visual
interpretation to be performed with relative ease.
Recently, a novel DNA fingerprinting strategy was described
independently by Welsh and McClelland (28) and Williams et
al. (29). A single oligonucleotide primer with an arbitrary
se-quence is used in a PCR to amplify and detect genetic DNA
polymorphisms and was called AP-PCR (28) or RAPD analysis
(29). Few studies have evaluated and compared the utility of
PCR-based methods for the typing of L. monocytogenes (7, 18).
Farber et al. (10) used three primers to test 52 L.
monocyto-genes strains of 11 serotypes and 12 strains belonging to 5 other
listerial species. RAPD analysis was able to subtype strains of
the same serotype and to differentiate between strains
belong-ing to different species. Only one primer found identical
pat-terns among strains of several different serotypes. Mazurier et
al. (18) found that RAPD analysis was comparable to
conven-tional phage typing but found RAPD analysis to be a quicker
and simpler method for epidemiologic studies. They
com-mented that not all laboratories are capable of performing
phage typing, and at present, a full complement of phages may
not be available to allow for the complete typeability of all
strains.
For AP-PCR typing, the selection of primers that produce
reproducible and easily interpretable DNA fingerprints is
es-sential. Unlike previous studies by RAPD analysis with short
primers (10 bases) (7, 10, 18, 19), the present study used longer
primers (17 and 18 bases) for amplification. Primers of 10
bases may produce fewer amplified products than longer
prim-ers (
$
18 bases) on the basis of the ‘‘context effect’’ (17). This
effect was observed in trial primer selection experiments with
shorter primers (data not shown). The use of 17- and 18-base
primers minimized this variation in the intensities of AP-PCR
products because of uneven amplification and produced
fin-gerprints that could be easily interpreted.
Although the results do show genotypic diversity in L.
mono-cytogenes strains when they are defined by AP-PCR
finger-prints and PFGE, further evaluation by studying the
distribu-tion of patterns among large and different populadistribu-tions of
Listeria serotypes and species is needed. The DI values for the
three typing methods were closely clustered, and as stated by
Hunter and Gaston (14), DI values must be regarded with
caution for small samples and typing schemes should not be
validated with limited sample sizes. Overall, the abilities of
AP-PCR and PFGE to differentiate between strains were
com-parable, and either method can be used to determine
related-ness or differences among L. monocytogenes strains.
Conven-TABLE 2. Number of types and DIs of three typing methods usedto type 51 strains of L. monocytogenes
Typing method No. of types DIa
RT with EcoRI 16 0.820
RT with PvuII 23 0.908
PFGE with ApaI 22 0.922
PFGE with SmaI 26 0.930
AP-PCR with PJ108 29 0.928
AP-PCR with PJ118 31 0.955
aThe DI was calculated by the following equation described by Hunter and Gaston (14):
D512N~N121!
O
j51 S
nj~nj21!
on May 15, 2020 by guest
http://jcm.asm.org/
[image:4.612.58.297.91.187.2]tional PFGE protocols may take up to 7 days or longer to
complete, although more rapid protocols are being described
(4). However, AP-PCR can be more rapid and easier to
per-form. Same-day results can be obtained if boiled cells are used
for template DNA in amplification reactions (unpublished
data; 16). As in other studies performed by using AP-PCR (10,
16, 18, 19), at least two independent primers should be used for
AP-PCR to improve its discriminatory power.
It is clear that no single method is sufficient for typing L.
monocytogenes. The choice of an optimal typing method in a
laboratory will depend on several factors. Ideally, the typing
method should be inexpensive, rapid, and simple to perform
with commonly available equipment. Swaminathan and Matar
(27) compared the costs of subtyping by RT-, PFGE-, or
PCR-based typing and found that PCR was the least costly per
sample; this was followed by PFGE. Many laboratories cannot
afford the start-up costs of an electrophoresis system for
PFGE, whereas many laboratories have invested in a
thermo-cyler, which can be used for both diagnostic and epidemiologic
purposes. Given these limitations, AP-PCR with at least two
different primers would be feasible for obtaining rapid results,
and when it is complemented with PFGE, it would provide
excellent discriminatory power.
ACKNOWLEDGMENTS
We thank Peter Jessamine and Anne Phillips for kindly providing some of the study isolates and Lisa Louie and Candy Rutherford for excellent technical expertise and assistance. We also gratefully ac-knowledge Kathryn Bernard and Judith Winstanley from the Special Bacteriology Section, Laboratory Centre for Disease Control, Ottawa, Ontario, Canada, for serotyping the Ontario strains of L.
monocyto-genes used in the study.
REFERENCES
1. Audurier, A., and C. Martin. 1989. Phage typing of Listeria monocytogenes. Int. J. Food Microbiol. 8:251–257.
2. Ausubel, F. M., R. Brent, R. E. Kingston, D. D. Moore, J. Seidman, J. A. Smith, and K. Struhl (ed.).1989. Current protocols in molecular biology. John Wiley & Sons, Inc., New York.
3. Baloga, A. O., and S. K. Harlander. 1991. Comparison of methods for discrimination between strains of Listeria monocytogenes from epidemiolog-ical surveys. Appl. Environ. Microbiol. 57:2324–2331.
4. Bannerman, T. L., G. A. Hancock, F. C. Tenover, and J. M. Miller. 1995. Pulsed-field gel electrophoresis as a replacement for bacteriophage typing of Staphylococcus aureus. J. Clin. Microbiol. 33:551–555.
5. Bibb, W. F., B. Schwartz, B. G. Gellin, B. D. Plikaytis, and R. E. Weaver. 1989. Analysis of Listeria monocytogenes by multilocus enzyme electrophore-sis and application of the method to epidemiologic investigations. Int. J. Food Microbiol. 8:233–239.
6. Bille, J., and M. P. Doyle. 1991. Listeria and Erysipelothrix, p. 287–295. In A. Balows, W. J. Hausler, Jr., K. L. Herrmann, H. D. Isenberg, and H. J. Shadomy (ed.), Manual of clinical microbiology, 5th ed. American Society for Microbiology, Washington, D.C.
7. Boerlin, P., E. Bannerman, F. Ischer, J. Rocourt, and J. Bille. 1995. Typing Listeria monocytogenes: a comparison of random amplification of polymor-phic DNA with 5 other methods. Res. Microbiol. 146:35–49.
8. Brosch, R., J. Chen, and J. B. Luchansky. 1994. Pulsed-field fingerprinting of listeriae: identification of genomic divisions for Listeria monocytogenes and their correlation with serovar. Appl. Environ. Microbiol. 60:2584–2592. 9. Brosius, J., A. Ulrich, M. A. Raker, A. Gray, T. J. Dull, R. R. Gutell, and
H. F. Noller.1981. Construction and fine mapping of recombinant plasmid
containing the rrnB ribosomal RNA operon of Escherichia coli. Plasmid 6:112–118.
10. Farber, J. M., and C. J. Addison. 1994. RAPD typing for distinguishing species and strains in the genus Listeria. J. Appl. Bacteriol. 77:242–250. 11. Farber, J. M., and P. I. Peterkin. 1991. Listeria monocytogenes, a food-borne
pathogen. Microbiol. Rev. 55:476–511.
12. Gellin, B. G., and C. V. Broome. 1989. Listeriosis. JAMA 261:1313–1320. 13. Graves, L. M., B. Swaminathan, M. W. Reeves, S. B. Hunter, R. E. Weaver,
B. D. Plikaytis, and A. Schuchat.1994. Comparison of ribotyping and mul-tilocus enzyme electrophoresis for subtyping of Listeria monocytogenes iso-lates. J. Clin. Microbiol. 32:2936–2943.
14. Hunter, P. R., and M. A. Gaston. 1988. Numerical index of the discrimina-tory ability of typing systems: an application of Simpson’s index of diversity. J. Clin. Microbiol. 26:2465–2466.
15. Linnan, M. J., L. Mascola, X. D. Lou, V. Goulet, S. May, C. Salminen, D. W. Hird, M. L. Yonekura, P. Hayes, R. Weaver, A. Audurier, B. D. Plikaytis, S. L. Fannin, A. Kleks, and C. V. Broome.1988. Epidemic listeriosis asso-ciated with Mexican-style cheese. N. Engl. J. Med. 319:823–828. 15a.Louie, M., J. Yao, P. Jayaratne, I. Luchsinger, J. Devenish, L. Louie, C.
Rutherford, W. Schlech, and A. E. Simor.1994. Molecular typing methods for the epidemiologic investigation of Listeria monocytogenes infection, abstr. J177, p. 171. In Program and abstracts of the 34th Interscience Conference on Antimicrobial Agents and Chemotherapy. American Society for Micro-biology, Washington, D.C.
16. MacGowan, A. P., K. O’Donaghue, S. Nicholls, J. McLauchlin, P. M. Ben-nett, and D. S. Reeves.1993. Typing of Listeria spp. by random amplified polymorphic DNA (RAPD) analysis. J. Med. Microbiol. 38:322–327. 17. Maslow, J. N., A. M. Slutsky, and R. D. Arbeit. 1993. Application of
pulsed-field gel electrophoresis for molecular epidemiology, p. 563–572. In D. H. Persing, T. F. Smith, F. C. Tenover, and T. J. White (ed.), Diagnostic molecular microbiology: principles and applications. American Society for Microbiology, Washington, D.C.
18. Mazurier, S. I., A. Audurier, N. Marquet-Van der Mee, S. Notermans, and K. Wernars.1992. A comparative study of randomly amplified polymorphic DNA analysis and conventional phage typing for epidemiological studies of Listeria monocytogenes isolates. Res. Microbiol. 143:507–512.
19. Mazurier, S. I., and K. Wernars. 1992. Typing of Listeria strains by random amplification of polymorphic DNA. Res. Microbiol. 143:499–505. 20. McLauchlin, J. 1990. Distribution of serovars of Listeria monocytogenes
isolated from different categories of patients with listeriosis. Eur. J. Clin. Microbiol. Infect. Dis. 9:210–213.
21. Nocera, D., M. Altwegg, G. M. Lucchini, E. Bannerman, F. Ischer, J. Rocourt, and J. Bille.1993. Characterization of Listeria strains from a food-borne listeriosis outbreak by rDNA gene restriction patterns compared to four other typing methods. Eur. J. Clin. Microbiol. Infect. Dis. 12:162–169. 22. Norrung, B., and P. Gerner-Smidt. 1993. Comparison of multilocus enzyme electrophoresis (MEE), ribotyping, restriction enzyme analysis (REA) and phage typing for typing of Listeria monocytogenes. Epidemiol. Infect. 111: 71–79.
23. Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
24. Schlech, W. F., P. M. Lavigne, R. A. Bortolussi, A. C. Allen, E. V. Haldane, A. J. Wort, A. W. Hightower, S. E. Johnson, S. H. King, E. S. Nicholls, and C. V. Broome.1983. Epidemic listeriosis—evidence for transmission by food. N. Engl. J. Med. 308:203–206.
25. Schuchat, A., B. Swaminathan, and C. V. Broome. 1991. Epidemiology of human listeriosis. Clin. Microbiol. Rev. 4:169–183.
26. Seeliger, H. P. R., and K. Hohne. 1979. Serotyping of Listeria monocytogenes and related species. Methods Microbiol. 13:31–49.
27. Swaminathan, B., and G. M. Matar. 1993. Molecular typing methods, p. 26–50. In D. H. Persing, T. F. Smith, F. C. Tenover, and T. J. White (ed.), Diagnostic molecular microbiology: principles and applications. American Society for Microbiology, Washington, D.C.
28. Welsh, J., and M. McClelland. 1990. Fingerprinting genomes using PCR with arbitrary primers. Nucleic Acids Res. 18:7213–7218.
29. Williams, J. G. K., A. R. Kubelik, K. J. Livak, J. A. Rafalski, and S. V. Tingey.1990. DNA polymorphisms amplified by arbitrary primers are useful as genetic markers. Nucleic Acids Res. 18:6531–6535.