Acta Cryst.(2003). E59, o799±o801 DOI: 10.1107/S160053680300998X Dominic LaliberteÂet al. C29H28O4
o799
organic papers
Acta Crystallographica Section E Structure Reports
Online
ISSN 1600-5368
1,3-Diphenoxy-2,2-bis(phenoxymethyl)propane
Dominic LaliberteÂ, Thierry Maris* and James D. Wuest
Universite de MontreÂal, DeÂpartement de Chimie, MontreÂal, QueÂbec, Canada H3C 3J7
Correspondence e-mail: [email protected]
Key indicators Single-crystal X-ray study
T= 220 K
Mean(C±C) = 0.003 AÊ
Rfactor = 0.050
wRfactor = 0.141 Data-to-parameter ratio = 8.5
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2003 International Union of Crystallography Printed in Great Britain ± all rights reserved
The title compound, C29H28O4, was synthesized by a new
method, crystallized from benzene/hexane, and its crystal structure determined. The molecule adopts a distorted tetrahedral geometry, and each phenyl group forms four edge-to-face phenyl±phenyl interactions with three different neighboring molecules.
Comment
Derivatives of tetraphenylmethane have been widely used as tetrahedral building blocks for molecular construction, leading to supramolecular networks, dendrimers, polymers, nanoscale structures, optoelectronic materials, liquid crystals, and other materials (Fournier, Maris et al., 2003; Fournier, Wanget al., 2003). The title compound, (I), provides a related subunit that is easier to make and more ¯exible. In the course of studying its use as a building block in supramolecular assembly, we investigated its structure to identify the preferred conformation, analyse the principal intermolecular interactions, and obtain detailed geometric information.
The title compound has been previously synthesized by the reaction of alkali salts of phenol with pentaerythrityl tetra-bromide (Backer & Dijken, 1936) or with pentaerythrityl tetratosylate (Shostakovskii et al., 1965). We made the compound by a modi®cation of the second route, obtained crystals from benzene/hexane, and determined the crystal structure (Figs. 1±3). Our data con®rm and signi®cantly re®ne the major features of a very early structural approximation, using crystals grown from benzene/petroleum ether (Bein-temaet al., 1935).
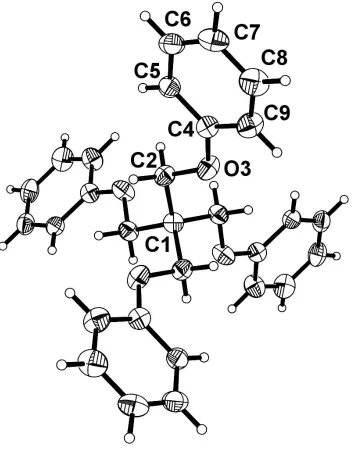
The two independent C2ÐC1ÐC20 angles at the central
atom C1, 108.58 (9) and 111.27 (19), are somewhat closer to
the ideal tetrahedral value than those of tetraphenylmethane, which are approximately 107 and 111(Robbinset al., 1975).
In addition, the arms connecting the central C atom to the phenyl groups are nearly fully extended, as shown by the torsion angle C1ÐC2ÐO3ÐC4 [175.05 (17)]. However, the
two independent C7ÐC1ÐC70angles de®ned by the central C
atom and the para positions of the phenyl groups have the values 87.77 (5) and 121.30 (3), showing that, overall, the
molecule deviates signi®cantly from tetrahedral geometry.
organic papers
o800
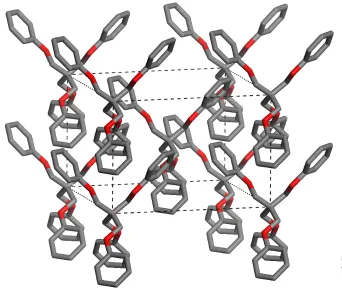
Dominic LaliberteÂet al. C29H28O4 Acta Cryst.(2003). E59, o799±o801 Cohesion in the crystal arises from van der Waals contactsand multiple edge-to-face phenyl±phenyl interactions. Each phenyl group participates in four of these interactions invol-ving three neighboring molecules. Two of the four phenyl± phenyl interactions de®ne part of a twofold embrace (Dance & Scudder, 1995), giving rise to chains along thecaxis (Fig. 3). In these embraces, the shortest H C distances (2.79 AÊ, with CÐH C angles of 150) are between the H atom attached to
C9 of one phenyl group and C6 of the other. The remaining two edge-to-face phenyl±phenyl interactions of each phenyl group involve neighbors in adjacent chains. In these interac-tions, the shortest H C distances (3.16 AÊ, with CÐH C angles of 133) are between the H atom attached to C6 of one
phenyl group and C5 of the other.
Experimental
Phenol (2.50 g, 26.6 mmol) and pentaerythrityl tetratosylate (4.00 g, 5.31 mmol) were added to Cs2CO3 (4.33 g, 13.3 mmol) inN,N0 -di-methylformamide (20 ml), and the mixture was heated at 413 K for 24 h. Water was then added, the resulting mixture was extracted with diethyl ether, and the organic phase was washed with water and dried over anhydrous MgSO4. Evaporation of solvent under reduced pressure left a residue which was ®ltered over silica gel, using chloroform as eluant, and then crystallized from benzene/hexane to give crystals of the title compound (1.25 g, 2.84 mmol, 53%).
Crystal data
C29H28O4 Mr= 440.51
Tetragonal,I4 a= 12.2242 (3) AÊ c= 8.4655 (3) AÊ V= 1265.01 (6) AÊ3 Z= 2
Dx= 1.156 Mg mÿ3
CuKradiation Cell parameters from 2339
re¯ections
= 5.1±69.7
= 0.61 mmÿ1 T= 220 (2) K Block, colorless 0.200.150.15 mm
Data collection
Bruker AXS SMART 2K/Platform diffractometer
!scans
Absorption correction: multi-scan (SADABS; Sheldrick, 1996) Tmin= 0.896,Tmax= 0.913 3443 measured re¯ections
643 independent re¯ections 613 re¯ections withI> 2(I) Rint= 0.059
max= 70.0 h=ÿ14!14 k=ÿ14!14 l=ÿ10!10
Figure 3
View of the chain parallel to thecaxis (vertical) generated by phenyl± phenyl interactions.
Figure 1
The structure of the title compound, with the atom-numbering scheme of the asymmetric unit. Displacement ellipsoids are drawn at the 30% probability level and H atoms are represented by spheres of arbitrary radius.
Figure 2
Re®nement
Re®nement onF2 R[F2> 2(F2)] = 0.050 wR(F2) = 0.141 S= 1.08 643 re¯ections 76 parameters
H-atom parameters constrained
w= 1/[2(F
o2) + (0.1128P)2
+ 0.0245P]
whereP= (Fo2+ 2Fc2)/3
(/)max< 0.001 max= 0.15 e AÊÿ3 min=ÿ0.20 e AÊÿ3
Extinction correction:SHELXL97 Extinction coef®cient: 0.013 (2)
Table 1
Selected geometric parameters (AÊ,). C1ÐC2 1.527 (2) C2ÐO3 1.419 (3) O3ÐC4 1.362 (3) C4ÐC5 1.386 (3) C4ÐC9 1.386 (3)
C5ÐC6 1.382 (4) C6ÐC7 1.373 (4) C7ÐC8 1.392 (5) C8ÐC9 1.371 (4)
C2iÐC1ÐC2 108.58 (9) C2ÐC1ÐC2ii 111.27 (19) O3ÐC2ÐC1 108.23 (15) C4ÐO3ÐC2 117.91 (16) O3ÐC4ÐC5 124.6 (2) O3ÐC4ÐC9 115.50 (19)
C5ÐC4ÐC9 119.9 (2) C6ÐC5ÐC4 119.2 (2) C7ÐC6ÐC5 121.4 (2) C6ÐC7ÐC8 119.0 (3) C9ÐC8ÐC7 120.4 (3) C8ÐC9ÐC4 120.2 (2)
C2iÐC1ÐC2ÐO3 61.53 (10) C2iiÐC1ÐC2ÐO3 ÿ57.91 (13) C2iiiÐC1ÐC2ÐO3 ÿ177.36 (18) C1ÐC2ÐO3ÐC4 175.05 (17) C2ÐO3ÐC4ÐC5 11.9 (3) C2ÐO3ÐC4ÐC9 ÿ169.3 (2) O3ÐC4ÐC5ÐC6 ÿ179.7 (2)
C9ÐC4ÐC5ÐC6 1.6 (4) C4ÐC5ÐC6ÐC7 ÿ0.5 (4) C5ÐC6ÐC7ÐC8 ÿ0.6 (4) C6ÐC7ÐC8ÐC9 0.6 (5) C7ÐC8ÐC9ÐC4 0.5 (5) O3ÐC4ÐC9ÐC8 179.6 (3) C5ÐC4ÐC9ÐC8 ÿ1.6 (4)
Symmetry codes: (i) 1y;ÿ1ÿx;ÿz; (ii)ÿx;ÿ2ÿy;z; (iii)ÿ1ÿy;xÿ1;ÿz.
As no elements heavier than oxygen are present, the Friedel pairs were merged and the absolute structure could not be determined. H atoms were placed in idealized positions with their isotropic displa-cement parameters ®xed to 1.2Ueqof the atoms to which they are bonded.
Data collection:SMART(Bruker, 1999); cell re®nement:SMART; data reduction: SAINT (Bruker, 1999); program(s) used to solve structure: SHELXS97 (Sheldrick, 1997); program(s) used to re®ne structure:SHELXL97 (Sheldrick, 1997); molecular graphics:XPin
SHELXTL (Bruker, 1997); software used to prepare material for publication:SHELXTL.
We are grateful to the Natural Sciences and Engineering Research Council of Canada, the MinisteÁre de l'EÂducation du QueÂbec, the Canada Foundation for Innovation, the Canada Research Chairs Program, and Merck Frosst for ®nancial support. In addition, acknowledgement is made to the donors of the Petroleum Research Fund, administered by the American Chemical Society, for support of this research.
References
Backer, H. J. & Dijken, G. (1936).Rec. Trav. Chim.55, 22±32.
Beintema, J., Terpstra, P. & Van Weerden, W. J. (1935).Rec. Trav. Chim.54, 627±630.
Bruker (1997). SHELXTL. Release 5.10. Bruker AXS Inc., Madison, Wisconsin, USA.
Bruker (1999).SMART(Release 5.059) andSAINT(Release 6.06). Bruker AXS Inc., Madison, Wisconsin, USA.
Dance, I. & Scudder, M. (1995).J. Chem. Soc. Chem. Commun.pp. 1039±1040. Fournier, J.-H., Maris, T., Wuest, J. D., Guo, W. & Galoppini, E. (2003).J. Am.
Chem. Soc.125, 1002±1006.
Fournier, J.-H., Wang, X. & Wuest, J. D. (2003)Can. J. Chem.In the press. Robbins, A., Jeffrey, G. A., Chesick, J. P., Donohue, J., Cotton, F. A., Frenz, B.
A. & Murillo, C. A. (1975).Acta Cryst.B31, 2395±2399.
Sheldrick, G. M. (1996).SADABS. Bruker AXS Inc., Madison, Wisconsin, USA.
Sheldrick, G. M. (1997). SHELXS97 and SHELXL97. University of GoÈttingen, Germany.
Shostakovskii, M. F., Atavin, A. S. & Mirskova, A. N. (1965).Zh. Obshch. Khim.35, 804±807.
supporting information
sup-1
Acta Cryst. (2003). E59, o799–o801
supporting information
Acta Cryst. (2003). E59, o799–o801 [doi:10.1107/S160053680300998X]
1,3-Diphenoxy-2,2-bis(phenoxymethyl)propane
Dominic Lalibert
é
, Thierry Maris and James D. Wuest
S1. Comment
Derivatives of tetraphenylmethane have been widely used as tetrahedral building blocks for molecular construction,
leading to supramolecular networks, dendrimers, polymers, nanoscale structures, optoelectronic materials, liquid crystals,
and other materials (Fournier, Maris et al., 2003; Fournier, Wang & Wuest, 2003). The title compound, (I), offers a
related subunit that is easier to make and more flexible. In the course of studying its use as a building block in
supramolecular assembly, we investigated its structure to identify the preferred conformation, analyze the principal
intermolecular interactions, and obtain detailed geometric information.
The title compound has been previously synthesized by the reaction of alkali salts of phenol with pentaerythrityl
tetrabromide (Backer & Dijken, 1936) or with pentaerythrityl tetratosylate (Shostakovskii et al., 1965). We made the
compound by a modification of the second route, obtained crystals from benzene/hexane, and determined their structure
(Figs. 1–3). Our data confirm and significantly refine the major features of a very early structural approximation, using
crystals grown from benzene/petroleum ether (Beintema et al., 1935).
The two independent C2—C1—C2 angles at the central atom C1, which are 108.52 (8) and 111.39 (16)°, are somewhat
closer to the ideal tetrahedral value than those of tetraphenylmethane, which are approximately 107 and 111° (Robbins et
al., 1975). In addition, the arms connecting the central C atom to the phenyl groups are nearly fully extended, as shown by the torsion angle C1—C2—O3—C4 [175.08 (15)°]. However, the two independent C7–C1–C7 angles defined by the
central C atom and the para positions of the phenyl groups have the values 87.77 (5) and 121.30 (3)°, showing that the
overall molecule deviates significantly from tetrahedral geometry.
Cohesion in the crystal arises from van der Waals contacts and multiple edge-to-face phenyl–phenyl interactions. Each
phenyl group participates in four of these interactions involving three neighboring molecules. Two of the four phenyl–
phenyl interactions define part of a twofold embrace (Dance & Scudder, 1995), giving rise to chains along the c axis (Fig.
3). In these embraces, the shortest H···C distances) 2.79 Å, with C–H···C angles of 150°) are between the H atom attached
to C9 of one phenyl group and C6 of the other. The remaining two edge-to-face phenyl–phenyl interactions of each
phenyl group involve neighbors in adjacent chains. In these interactions, the shortest H···C distances (3.16 Å, with C–
H···C angles of 133°) are between the H atom attached to C6 of one phenyl group and C5 of the other.
S2. Experimental
Phenol (2.50 g, 26.6 mmol) and pentaerythrityl tetratosylate (4.00 g, 5.31 mmol) were added to Cs2CO3 (4.33 g, 13.3
mmol) in N,N′-dimethylformamide (20 ml), and the mixture was heated at 413 K for 24 h. Water was then added, the
resulting mixture was extracted with diethyl ether, and the organic phase was washed with water and dried over
anhydrous MgSO4. Evaporation of solvent under reduced pressure left a residue which was filtered over silica gel using
chloroform as eluent and then crystallized from benzene/hexane to give crystals of the title coupound (1.25 g, 2.84 mmol,
supporting information
sup-2
Acta Cryst. (2003). E59, o799–o801
S3. Refinement
As no atom types with Z > Si are present, the Friedel pairs were merged and the absolute structure could not be defined.
H atoms were rided to idealized positions with their isotropic displacement parameters fixed to 1.2 Ueq of the equivalent
[image:5.610.132.486.136.585.2]isotropic displacement parameter of the atoms to which they are bonded.
Figure 1
Displacement ellispsoids drawing of the structure of the title compound with the atom-numbering scheme. Ellipsoids are
supporting information
sup-3
[image:6.610.124.466.70.366.2]Acta Cryst. (2003). E59, o799–o801
Figure 2
supporting information
sup-4
[image:7.610.230.381.71.524.2]Acta Cryst. (2003). E59, o799–o801
Figure 3
View of the chain parallel to the c axis (vertical) generated by phenyl–phenyl interactions.
1,3-diphenoxy-2,2-bis(phenoxymethyl)propane
Crystal data C29H28O4 Mr = 440.51 Tetragonal, I4 Hall symbol: I -4 a = 12.2242 (3) Å c = 8.4655 (3) Å V = 1265.01 (6) Å3 Z = 2
F(000) = 468
Dx = 1.156 Mg m−3 Melting point: 113 K
Cu Kα radiation, λ = 1.54178 Å Cell parameters from 2339 reflections θ = 5.1–69.7°
supporting information
sup-5
Acta Cryst. (2003). E59, o799–o801 Data collection
Bruker AXS SMART 2K/Platform diffractometer
Radiation source: Sealed Tube Graphite monochromator
Detector resolution: 5.5 pixels mm-1 ω scans
Absorption correction: multi-scan (SADABS; Sheldrick, 1996) Tmin = 0.896, Tmax = 0.913
3443 measured reflections 643 independent reflections 613 reflections with I > 2σ(I) Rint = 0.059
θmax = 70.0°, θmin = 5.1° h = −14→14
k = −14→14 l = −10→10
Refinement Refinement on F2 Least-squares matrix: full R[F2 > 2σ(F2)] = 0.050 wR(F2) = 0.141 S = 1.08 643 reflections 76 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H-atom parameters constrained w = 1/[σ2(F
o2) + (0.1128P)2 + 0.0245P] where P = (Fo2 + 2Fc2)/3
(Δ/σ)max < 0.001 Δρmax = 0.15 e Å−3 Δρmin = −0.20 e Å−3
Extinction correction: SHELXL97, Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4 Extinction coefficient: 0.013 (2)
Special details
supporting information
sup-6
Acta Cryst. (2003). E59, o799–o801
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Angle CCC1
87.77 (0.05) C7 - C1 - C7_$1 Angle CCC2
121.30 (0.03) C7 - C1 - C7_$2 Angle CCC3
121.30 (0.03) C7 - C1 - C7_$3 Distance LH1
2.7845 (0.036) C6 - H9_$4 Angle ALH1
150.69 (2.93) C6 - H9_$4 - C9_$4 Distance LH2
3.1582 (0.036) H6 - C5_$5 Angle ALH2
133.41 (2.55) C6 - H6 - C5_$5
Least-squares planes (x,y,z in crystal coordinates) and deviations from them (* indicates atom used to define plane) 7.0504 (0.0120) x − 6.9471 (0.0128) y − 4.9678 (0.0073) z = 6.6087 (0.0137)
* 0.0093 (0.0017) C4 * −0.0056 (0.0016) C5 * −0.0020 (0.0017) C6 * 0.0059 (0.0020) C7 * −0.0021 (0.0023) C8 * −0.0055 (0.0020) C9
Rms deviation of fitted atoms = 0.0056
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
C1 0.0000 −1.0000 0.0000 0.0504 (10)
C2 −0.10139 (18) −1.01889 (18) −0.1018 (3) 0.0523 (7)
H2A −0.0921 −1.0850 −0.1659 0.063*
H2B −0.1659 −1.0285 −0.0345 0.063*
O3 −0.11545 (14) −0.92649 (13) −0.2012 (2) 0.0592 (6)
C4 −0.19684 (17) −0.93000 (18) −0.3110 (3) 0.0483 (6)
C5 −0.27891 (17) −1.00814 (18) −0.3152 (3) 0.0522 (6)
H5 −0.2816 −1.0638 −0.2387 0.063*
C6 −0.35678 (18) −1.0033 (2) −0.4333 (4) 0.0619 (7)
H6 −0.4122 −1.0565 −0.4368 0.074*
C7 −0.3549 (2) −0.9222 (3) −0.5455 (4) 0.0687 (8)
H7 −0.4080 −0.9202 −0.6258 0.082*
C8 −0.2732 (3) −0.8431 (3) −0.5386 (4) 0.0726 (9)
H8 −0.2714 −0.7868 −0.6142 0.087*
C9 −0.1953 (2) −0.8468 (2) −0.4222 (4) 0.0647 (8)
H9 −0.1407 −0.7927 −0.4178 0.078*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
supporting information
sup-7
Acta Cryst. (2003). E59, o799–o801
C2 0.0533 (11) 0.0528 (11) 0.0507 (14) −0.0065 (8) −0.0010 (11) 0.0035 (10)
O3 0.0704 (10) 0.0584 (9) 0.0486 (11) −0.0173 (7) −0.0122 (9) 0.0092 (8)
C4 0.0528 (10) 0.0554 (11) 0.0365 (12) −0.0014 (8) 0.0037 (10) −0.0021 (10)
C5 0.0501 (10) 0.0524 (11) 0.0541 (13) 0.0026 (7) 0.0055 (11) −0.0034 (11)
C6 0.0460 (10) 0.0712 (14) 0.0686 (18) 0.0037 (9) −0.0043 (12) −0.0082 (13)
C7 0.0530 (12) 0.0950 (18) 0.0580 (16) 0.0104 (11) −0.0062 (11) −0.0024 (15)
C8 0.0746 (15) 0.0926 (18) 0.0507 (16) 0.0053 (13) 0.0021 (13) 0.0196 (15)
C9 0.0676 (14) 0.0759 (15) 0.0506 (16) −0.0111 (11) 0.0013 (12) 0.0140 (12)
Geometric parameters (Å, º)
C1—C2 1.527 (2) C5—C6 1.382 (4)
C1—C2i 1.527 (2) C5—H5 0.94
C1—C2ii 1.527 (2) C6—C7 1.373 (4)
C1—C2iii 1.527 (2) C6—H6 0.94
C2—O3 1.419 (3) C7—C8 1.392 (5)
C2—H2a 0.98 C7—H7 0.94
C2—H2b 0.98 C8—C9 1.371 (4)
O3—C4 1.362 (3) C8—H8 0.94
C4—C5 1.386 (3) C9—H9 0.94
C4—C9 1.386 (3)
C2i—C1—C2 108.58 (9) C6—C5—C4 119.2 (2)
C2i—C1—C2ii 108.58 (9) C6—C5—H5 120.4
C2—C1—C2ii 111.27 (19) C4—C5—H5 120.4
C2i—C1—C2iii 111.27 (19) C7—C6—C5 121.4 (2)
C2—C1—C2iii 108.58 (9) C7—C6—H6 119.3
C2ii—C1—C2iii 108.58 (9) C5—C6—H6 119.3
O3—C2—C1 108.23 (15) C6—C7—C8 119.0 (3)
O3—C2—H2A 110.1 C6—C7—H7 120.5
C1—C2—H2A 110.1 C8—C7—H7 120.5
O3—C2—H2B 110.1 C9—C8—C7 120.4 (3)
C1—C2—H2B 110.1 C9—C8—H8 119.8
H2A—C2—H2B 108.4 C7—C8—H8 119.8
C4—O3—C2 117.91 (16) C8—C9—C4 120.2 (2)
O3—C4—C5 124.6 (2) C8—C9—H9 119.9
O3—C4—C9 115.50 (19) C4—C9—H9 119.9
C5—C4—C9 119.9 (2)
C2i—C1—C2—O3 61.53 (10) C9—C4—C5—C6 1.6 (4)
C2ii—C1—C2—O3 −57.91 (13) C4—C5—C6—C7 −0.5 (4)
C2iii—C1—C2—O3 −177.36 (18) C5—C6—C7—C8 −0.6 (4)
C1—C2—O3—C4 175.05 (17) C6—C7—C8—C9 0.6 (5)
C2—O3—C4—C5 11.9 (3) C7—C8—C9—C4 0.5 (5)
C2—O3—C4—C9 −169.3 (2) O3—C4—C9—C8 179.6 (3)
O3—C4—C5—C6 −179.7 (2) C5—C4—C9—C8 −1.6 (4)