organic papers
Acta Cryst.(2005). E61, o2005–o2007 doi:10.1107/S1600536805017290 Yao-Cheng Shi C
12H15NO2
o2005
Acta Crystallographica Section E Structure Reports Online
ISSN 1600-5368
R
2 2(14) dimers in
3-(hydroxyethyl)amino-1-phenylbut-2-en-1-one
Yao-Cheng Shi
School of Chemistry, Yangzhou University, 130 XiMenWai Street, Yangzhou 225002, People’s Republic of China
Correspondence e-mail: yzssyc@yzcn.net
Key indicators
Single-crystal X-ray study
T= 293 K
Mean(C–C) = 0.005 A˚
Rfactor = 0.071
wRfactor = 0.220
Data-to-parameter ratio = 16.2
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2005 International Union of Crystallography
Printed in Great Britain – all rights reserved
The title compound, C6H5COCH C(NHCH2CH2OH)CH3
or C12H15NO2, has been synthesized by the reaction of
benzoylacetone and ethanolamine in ethanol. In the crystal-line state, molecules are linked by intermolecular hydrogen
bonds (O—H O C) to form centrosymmetric R2
2
(14) dimers.
Comment
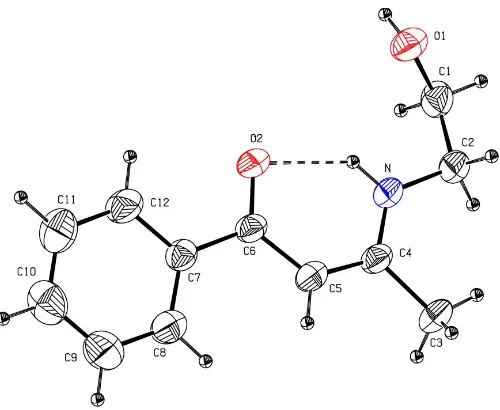
The title compound, (I), has been synthesized as part of an ongoing investigation of the chemistry of enaminones (Shi, 2005a,b,c) and its crystal structure has been determined (Fig. 1). An organometallic analog of (I), hereafter (II), has already been studied (Shiet al., 2005). The bond lengths and angles of the O C—C C—N system in (I) are similar to the corresponding values in (II) (Table 1). For the molecules of (I) and (II), the bond lengths in the O C—C C—N system indicate electron delocalization (Shi, Yang, Shenet al., 2004; Shi, Yang, Song & Liu, 2004; Gilliet al., 2000; Gilchrist, 1997). The dihedral angle between the O C—C C—N system and the aromatic ring in (I) is 8.3 (2); the value in (II) is 14.8 (2). Therefore, the above dihedral angles suggest that the benzene ring in (I) and the substituted cyclopentadienyl ring in (II) are
not involved in the conjugation of the O C—C C—N
system; this is in agreement with the fact that the bonds linking the O C—C C—N system and the aromatic ring in (I) and (II) are typical single bonds.
As in (II), the strong intramolecular hydrogen bond involving the enamine N and carbonyl O atom in (I) stabilizes
the enaminone, whereas intermolecular O—H O C
hydrogen bonds form centrosymmetricR2
2(14) dimers
(Bern-steinet al., 1995; Table 2 and Fig. 2). Interestingly, non-classical C—H O C hydrogen bonds are present in the crystalline state of (I), whereas the corresponding intermolecular hydrogen bonds in (II) are not observed.
Experimental
The title compound was synthesized by refluxing a solution of benzoylacetone and ethanolamine (1:1) in ethanol for 4 h. It was
recrystallized from CH2Cl2–petroleum ether as colorless crystals
(m.p. 359–361 K). IR (KBr): 3332 (br, s, NH, OH), 1602 (vs, C O), 1532 (vs, C C) cm1.H NMR (600 MHz, CDCl3):11.498 (1H,s,
NH), 7.838, 7.395 (2H,m, 3 H,m, C6H5), 5.663 (1H,s, CH), 3.814 (2H,
t,3J= 5.4 Hz, OCH2), 3.492 (2H,t, 3
J= 5.4 Hz, NCH2), 2.881 (br, 1H,
OH), 2.032 (3H,s, CH3).
Crystal data
C12H15NO2
Mr= 205.25 Monoclinic,P21=n
a= 5.6330 (11) A˚
b= 25.961 (5) A˚
c= 7.9770 (16) A˚
= 102.26 (3)
V= 1139.9 (4) A˚3
Z= 4
Dx= 1.196 Mg m
3 MoKradiation Cell parameters from 25
reflections
= 10–13
= 0.08 mm1
T= 293 K Block, Colorless 0.40.30.2 mm
Data collection
Enraf–Nonius CAD-4 diffractometer
!/2scans
Absorption correction: scan (Northet al., 1968)
Tmin= 0.955,Tmax= 0.976 2449 measured reflections 2224 independent reflections 1044 reflections withI> 2(I)
Rint= 0.050
max= 26.0
h= 0!6
k= 0!31
l=9!9 3 standard reflections
every 200 reflections intensity decay: none
Refinement
Refinement onF2 R[F2> 2(F2)] = 0.071
wR(F2) = 0.220
S= 0.97 2224 reflections 137 parameters
H-atom parameters constrained
w= 1/[2
(Fo2) + (0.08P)2 + 0.8P]
whereP= (Fo2+ 2Fc2)/3 (/)max< 0.001
max= 0.32 e A˚
3
min=0.20 e A˚
3
Extinction correction:SHELXL97
Extinction coefficient: 0.065 (8)
Table 1
Selected geometric parameters (A˚ ,).
N—C2 1.449 (4) N—C4 1.318 (4) O1—C1 1.426 (4) O2—C6 1.254 (4) C1—C2 1.490 (5)
C3—C4 1.512 (4) C4—C5 1.386 (5) C5—C6 1.414 (4) C6—C7 1.496 (5)
C2—N—C4 127.0 (3) O1—C1—C2 109.5 (3) N—C2—C1 109.3 (3) N—C4—C3 118.2 (3) N—C4—C5 121.7 (3)
C3—C4—C5 120.0 (3) C4—C5—C6 124.2 (3) O2—C6—C5 122.2 (3) O2—C6—C7 118.2 (3) C5—C6—C7 119.6 (3)
Table 2
Hydrogen-bond geometry (A˚ ,).
D—H A D—H H A D A D—H A
N—Hn O2 0.86 1.98 2.658 (4) 135 O1—Ho O2i
0.82 1.93 2.747 (4) 179 C3—H3b O2ii
0.96 2.54 3.443 (5) 158
Symmetry codes: (i)xþ2;y;zþ1; (ii)x1;y;z.
All H atoms were located in difference maps and subsequently treated as riding atoms, with C—H = 0.93 (olefinic and aromatic), 0.96 (methyl) or 0.97 A˚ (methylene), N—H = 0.86 A˚ and O—H = 0.82 A˚.
Uiso(H) values were set at 1.2Ueq(C,N), 1.5Ueq(Cmethyl) or 1.5Ueq(O).
Data collection: CAD-4 Software (Enraf–Nonius, 1989); cell refinement: CAD-4 Software; data reduction: XCAD4 (Harms & Wocadlo, 1995); program(s) used to solve structure: SHELXS97
(Sheldrick, 1997); program(s) used to refine structure:SHELXL97
(Sheldrick, 1997); molecular graphics: ORTEP-3 for Windows
(Farrugia, 1997); software used to prepare material for publication:
PLATON(Spek, 2003).
The author thanks Yangzhou University for financial support.
organic papers
o2006
Yao-Cheng Shi C [image:2.610.316.566.68.273.2]12H15NO2 Acta Cryst.(2005). E61, o2005–o2007
Figure 1
[image:2.610.313.566.328.597.2]The molecular structure of (I). Displacement ellipsoids for non-H atoms are drawn at the 30% probability level. The intramolecular hydrogen bond is shown as a dashed line.
Figure 2
References
Bernstein, J., Davis, R. E., Shimoni, L. & Chang, N.-L. (1995).Angew. Chem. Int. Ed. Engl.34, 1555–1573.
Enraf–Nonius (1989). CAD-4 Software. Enraf–Nonius, Delft, The Nether-lands.
Farrugia, L. J. (1997).J. Appl. Cryst.30, 565.
Gilchrist, T. L. (1997). Heterocyclic Chemistry, 3rd ed. London: Addison Wesley Longman Limited.
Gilli, P., Bertolasi, V., Ferretti, V. & Gilli, G. (2000).J. Am. Chem. Soc.122, 10405–10412.
Harms, K. & Wocadlo, S. (1995).XCAD4. University of Marburg, Germany. North, A. C. T., Phillips, D. C. & Mathews, F. S. (1968).Acta Cryst.A24, 351–
359.
Sheldrick, G. M. (1997). SHELXS97 and SHELXL97. University of Go¨ttingen, Germany.
Shi, Y.-C. (2005a).Acta Cryst.E61, m811–m812. Shi, Y.-C. (2005b).Acta Cryst.E61, m873–m874. Shi, Y.-C. (2005c).Acta Cryst.E61, o1130–o1132.
Shi, Y.-C., Yang, H.-M., Shen, W.-B., Yan, C.-G. & Hu, X.-Y. (2004).
Polyhedron,23, 15–21.
Shi, Y.-C., Yang, H.-M., Song, H.-B. & Liu, Y.-H. (2004).Polyhedron,23, 1541– 1546.
Shi, Y.-C., Sui, C.-X., Song, H.-B. & Jian, P.-M. (2005).J. Coord. Chem.58, 363– 371.
Spek, A. L. (2003).J. Appl. Cryst.36, 7–13.
organic papers
Acta Cryst.(2005). E61, o2005–o2007 Yao-Cheng Shi C
supporting information
sup-1 Acta Cryst. (2005). E61, o2005–o2007
supporting information
Acta Cryst. (2005). E61, o2005–o2007 [https://doi.org/10.1107/S1600536805017290]
R
22(14) dimers in 3-(hydroxyethyl)amino-1-phenylbut-2-en-1-one
Yao-Cheng Shi
3-(Hydroxyethylamino)-1-phenylbut-2-en-1-one
Crystal data
C12H15NO2
Mr = 205.25
Monoclinic, P21/n Hall symbol: -P 2yn
a = 5.6330 (11) Å
b = 25.961 (5) Å
c = 7.9770 (16) Å
β = 102.26 (3)°
V = 1139.9 (4) Å3
Z = 4
F(000) = 440
Dx = 1.196 Mg m−3
Mo Kα radiation, λ = 0.71073 Å
Cell parameters from 25 reflections
θ = 10–13°
µ = 0.08 mm−1
T = 293 K
Block, colorless 0.4 × 0.3 × 0.2 mm
Data collection
Enraf–Nonius CAD-4 diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
ω/2θ scans
Absorption correction: ψ scan
(North et al., 1968)
Tmin = 0.955, Tmax = 0.976 2449 measured reflections
2224 independent reflections 1044 reflections with I > 2σ(I)
Rint = 0.050
θmax = 26.0°, θmin = 1.6°
h = 0→6
k = 0→31
l = −9→9
3 standard reflections every 200 reflections intensity decay: none
Refinement
Refinement on F2
Least-squares matrix: full
R[F2 > 2σ(F2)] = 0.071
wR(F2) = 0.220
S = 0.97
2224 reflections 137 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H-atom parameters constrained
w = 1/[σ2(F
o2) + (0.08P)2 + 0.8P] where P = (Fo2 + 2Fc2)/3 (Δ/σ)max < 0.001
Δρmax = 0.32 e Å−3 Δρmin = −0.20 e Å−3
supporting information
sup-2 Acta Cryst. (2005). E61, o2005–o2007
Special details
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only
used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and
goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of
reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors
based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
O1 1.0052 (4) −0.01119 (10) 0.7678 (3) 0.0753 (9)
Ho 1.0553 −0.0344 0.7150 0.113*
O2 0.8199 (4) 0.08835 (10) 0.4083 (3) 0.0713 (8)
N 0.6354 (5) 0.06123 (12) 0.6757 (3) 0.0616 (9)
Hn 0.7361 0.0550 0.6109 0.074*
C1 0.7635 (7) −0.02264 (16) 0.7842 (5) 0.0720 (11)
H1a 0.7676 −0.0465 0.8784 0.086*
H1b 0.6750 −0.0388 0.6798 0.086*
C2 0.6388 (7) 0.02580 (15) 0.8167 (5) 0.0675 (11)
H2a 0.4739 0.0182 0.8270 0.081*
H2b 0.7239 0.0413 0.9232 0.081*
C3 0.3060 (7) 0.11262 (17) 0.7411 (5) 0.0793 (13)
H3a 0.3168 0.0869 0.8290 0.119*
H3b 0.1470 0.1117 0.6680 0.119*
H3c 0.3344 0.1460 0.7931 0.119*
C4 0.4946 (6) 0.10189 (15) 0.6362 (4) 0.0582 (9)
C5 0.5131 (6) 0.13388 (14) 0.5004 (5) 0.0620 (10)
H5 0.4147 0.1630 0.4821 0.074*
C6 0.6720 (6) 0.12509 (14) 0.3879 (4) 0.0555 (9)
C7 0.6630 (6) 0.15994 (14) 0.2374 (4) 0.0574 (9)
C8 0.4894 (8) 0.19711 (18) 0.1884 (6) 0.0915 (14)
H8 0.3719 0.2017 0.2532 0.110*
C9 0.4824 (10) 0.2278 (2) 0.0470 (7) 0.1073 (17)
H9 0.3641 0.2532 0.0199 0.129*
C10 0.6466 (10) 0.2214 (2) −0.0533 (7) 0.0989 (16)
H10 0.6392 0.2414 −0.1510 0.119*
C11 0.8222 (9) 0.1850 (2) −0.0079 (7) 0.1088 (18)
H11 0.9380 0.1809 −0.0744 0.131*
C12 0.8329 (7) 0.15384 (17) 0.1353 (6) 0.0819 (13)
supporting information
sup-3 Acta Cryst. (2005). E61, o2005–o2007
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
O1 0.0596 (15) 0.0871 (19) 0.0840 (19) 0.0027 (14) 0.0257 (13) −0.0078 (15)
O2 0.0566 (15) 0.0903 (19) 0.0739 (17) 0.0182 (15) 0.0296 (12) 0.0108 (14)
N 0.0498 (17) 0.083 (2) 0.0566 (18) 0.0056 (16) 0.0218 (14) 0.0040 (16)
C1 0.066 (2) 0.088 (3) 0.066 (2) −0.008 (2) 0.0216 (19) 0.009 (2)
C2 0.056 (2) 0.090 (3) 0.060 (2) 0.003 (2) 0.0208 (18) 0.007 (2)
C3 0.068 (3) 0.104 (3) 0.075 (3) 0.013 (2) 0.037 (2) −0.005 (2)
C4 0.0433 (19) 0.074 (2) 0.060 (2) 0.0006 (18) 0.0177 (16) −0.0075 (19)
C5 0.058 (2) 0.067 (2) 0.065 (2) 0.0073 (18) 0.0236 (18) −0.0034 (19)
C6 0.0419 (18) 0.066 (2) 0.060 (2) 0.0010 (18) 0.0142 (15) −0.0053 (18)
C7 0.050 (2) 0.065 (2) 0.059 (2) −0.0053 (18) 0.0148 (17) −0.0045 (18)
C8 0.095 (3) 0.098 (3) 0.091 (3) 0.034 (3) 0.043 (3) 0.020 (3)
C9 0.118 (4) 0.100 (4) 0.107 (4) 0.028 (3) 0.032 (3) 0.035 (3)
C10 0.096 (4) 0.099 (4) 0.102 (4) −0.020 (3) 0.023 (3) 0.036 (3)
C11 0.094 (4) 0.138 (5) 0.109 (4) −0.012 (4) 0.054 (3) 0.036 (4)
C12 0.065 (3) 0.093 (3) 0.097 (3) 0.005 (2) 0.038 (2) 0.017 (3)
Geometric parameters (Å, º)
N—C2 1.449 (4) C4—C5 1.386 (5)
N—C4 1.318 (4) C5—C6 1.414 (4)
N—Hn 0.8600 C5—H5 0.9300
O1—C1 1.426 (4) C6—C7 1.496 (5)
O1—Ho 0.8200 C7—C8 1.370 (5)
O2—C6 1.254 (4) C7—C12 1.391 (5)
C1—C2 1.490 (5) C8—C9 1.375 (6)
C1—H1a 0.9700 C8—H8 0.9300
C1—H1b 0.9700 C9—C10 1.355 (7)
C2—H2a 0.9700 C9—H9 0.9300
C2—H2b 0.9700 C10—C11 1.360 (6)
C3—C4 1.512 (4) C10—H10 0.9300
C3—H3a 0.9600 C11—C12 1.391 (6)
C3—H3b 0.9600 C11—H11 0.9300
C3—H3c 0.9600 C12—H12 0.9300
C1—O1—Ho 109.5 C4—C5—C6 124.2 (3)
C2—N—C4 127.0 (3) C4—C5—H5 117.9
C2—N—Hn 116.5 C6—C5—H5 117.9
C4—N—Hn 116.5 O2—C6—C5 122.2 (3)
O1—C1—C2 109.5 (3) O2—C6—C7 118.2 (3)
O1—C1—H1a 109.8 C5—C6—C7 119.6 (3)
C2—C1—H1a 109.8 C8—C7—C12 116.8 (4)
O1—C1—H1b 109.8 C8—C7—C6 123.7 (3)
C2—C1—H1b 109.8 C12—C7—C6 119.4 (3)
H1a—C1—H1b 108.2 C7—C8—C9 122.4 (4)
supporting information
sup-4 Acta Cryst. (2005). E61, o2005–o2007
N—C2—H2a 109.8 C9—C8—H8 118.8
C1—C2—H2a 109.8 C10—C9—C8 120.5 (5)
N—C2—H2b 109.8 C10—C9—H9 119.7
C1—C2—H2b 109.8 C8—C9—H9 119.7
H2a—C2—H2b 108.3 C9—C10—C11 118.5 (4)
N—C4—C3 118.2 (3) C9—C10—H10 120.7
N—C4—C5 121.7 (3) C11—C10—H10 120.7
C3—C4—C5 120.0 (3) C10—C11—C12 121.7 (4)
C4—C3—H3a 109.5 C10—C11—H11 119.2
C4—C3—H3b 109.5 C12—C11—H11 119.2
H3a—C3—H3b 109.5 C11—C12—C7 120.0 (4)
C4—C3—H3c 109.5 C11—C12—H12 120.0
H3a—C3—H3c 109.5 C7—C12—H12 120.0
H3b—C3—H3c 109.5
C4—N—C2—C1 162.3 (3) O2—C6—C7—C12 −6.3 (5)
O1—C1—C2—N 59.0 (4) C5—C6—C7—C12 174.7 (3)
C2—N—C4—C5 178.1 (3) C12—C7—C8—C9 −0.9 (7)
C2—N—C4—C3 −3.1 (5) C6—C7—C8—C9 −178.5 (4)
N—C4—C5—C6 3.2 (6) C7—C8—C9—C10 1.7 (8)
C3—C4—C5—C6 −175.6 (3) C8—C9—C10—C11 −2.0 (8)
C4—C5—C6—O2 −3.8 (6) C9—C10—C11—C12 1.6 (8)
C4—C5—C6—C7 175.2 (3) C10—C11—C12—C7 −0.8 (8)
O2—C6—C7—C8 171.2 (4) C8—C7—C12—C11 0.5 (6)
C5—C6—C7—C8 −7.9 (5) C6—C7—C12—C11 178.1 (4)
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
N—Hn···O1 0.86 2.45 2.786 (4) 104
N—Hn···O2 0.86 1.98 2.658 (4) 135
O1—Ho···O2i 0.82 1.93 2.747 (4) 179
C3—H3b···O2ii 0.96 2.54 3.443 (5) 158