organic papers
o614
Xiao-Qing Caiet al. C8H12N22C6H5NO3 doi:10.1107/S160053680500382X Acta Cryst.(2005). E61, o614–o615
Acta Crystallographica Section E Structure Reports Online
ISSN 1600-5368
2,3,5,6-Tetramethylpyrazine–
p
-nitrophenol (1/2)
Xiao-Qing Cai, Mao-Lin Hu,* Ya-Juan Zhao, Ya-Qian Cheng and Shun Wang
School of Chemistry and Materials Science, Wenzhou Normal College, Zhejiang, Wenzhou 325027, People’s Republic of China
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study
T= 298 K
Mean(C–C) = 0.004 A˚
Rfactor = 0.077
wRfactor = 0.170
Data-to-parameter ratio = 13.1
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2005 International Union of Crystallography Printed in Great Britain – all rights reserved
In the title complex, C8H12N22C6H5NO3, one
2,3,5,6-tetra-methylpyrazine and two p-nitrophenol molecules form a
centrosymmetric unit. Intermolecular O—H N and weak
C—H O hydrogen bonds are observed in the structure,
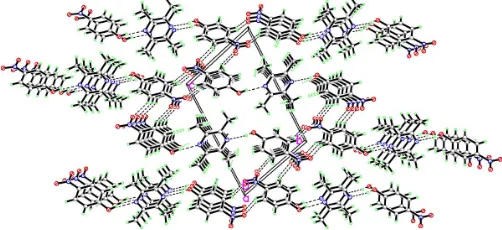
leading to the formation of a two-dimensional network.
Comment
2,3,5,6-Tetramethylpyrazine (TMP), which possesses anti-ulcer and antiplatelet activities, is a biologically active ingre-dient isolated from the traditional herbal medicineLigusticum chuanxiong Hort, widely used in China for the clinical treat-ment of nephrosis and ischemic cerebrovascular disease, due to its function of anticoagulation and angiectasis (Penget al., 2004). It also has a protective effect against ischemic neuronal
damage in the hippocampus (Su et al., 2004). p-Nitrophenol
(PNP), which is commonly used as a pesticide in agriculture, is a precursor of pharmaceuticals such as acetaminophen and
4-aminosalicylic acid (Kam et al., 2005). In the past, some
supramolecular compounds were synthesized with either TMP (Tian & Yang, 1993; Smythet al., 1996; Donget al., 2003) or PNP (Paternostreet al., 1998; Nget al., 2001). We report here the structure of the title compound, (I), containing both TMP and PNP.
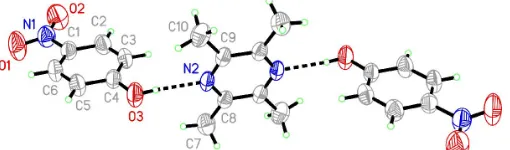
In (I), one TMP molecule links two PNP moleculesviaO—
H N hydrogen-bond interactions to form a centrosymmetric
unit (Fig. 1). The N2—C8, N2—C9, C9—C8i distances
[1.339 (3)–1.402 (3) A˚ ; symmetry code: (i) 1x, y, 1z] are between double- and single-bond lengths, indicating
[image:1.610.212.454.452.540.2] [image:1.610.207.462.636.711.2]Received 26 January 2005 Accepted 2 February 2005 Online 12 February 2005
Figure 1
electron delocalization in the pyrazine ring (Table 1). The dihedral angle between the pyrazine ring of TMP and the
benzene ring of PNP is 82.38 (9). The weak intermolecular
C—H O hydrogen bonds (Table 2) link the centrosymmetric
units into a two-dimensional network (Fig. 2).
Experimental
The title compound was obtained by the hydrothermal method from a mixture of 2,3,5,6-tetramethylpyrazine (2 mmol, 0.27 g) and p -nitrophenol (2 mmol, 0.28 g) in a 30 ml Teflon-lined stainless steel reactor. The solution was heated to 412 K for 3 d. On completion of the reaction, the system was cooled slowly to room temperature and the colorless block-shaped crystals which formed were collected.
Crystal data
C8H12N22C6H5NO3
Mr= 414.42
Triclinic,P1
a= 4.7521 (2) A˚
b= 8.8700 (4) A˚
c= 13.1941 (6) A˚ = 73.916 (2)
= 81.771 (2)
= 74.706 (2)
V= 513.98 (4) A˚3
Z= 1
Dx= 1.339 Mg m 3
MoKradiation Cell parameters from 1276
reflections = 2.5–25.2
= 0.10 mm1
T= 298 (2) K Block, colorless 0.480.350.15 mm
Data collection
Bruker SMART APEX area-detector diffractometer ’and!scans
Absorption correction: multi-scan (SADABS; Bruker, 2002)
Tmin= 0.953,Tmax= 0.985
2747 measured reflections
1825 independent reflections 1720 reflections withI> 2(I)
Rint= 0.012
max= 25.2
h=5!5
k=8!10
l=15!15
Refinement
Refinement onF2
R[F2> 2(F2)] = 0.077
wR(F2) = 0.170
S= 1.27 1825 reflections 139 parameters
H-atom parameters constrained
w= 1/[2(F
o2) + (0.0532P)2
+ 0.2196P]
whereP= (Fo2+ 2Fc2)/3
(/)max< 0.001 max= 0.16 e A˚
3
[image:2.610.45.296.74.189.2]min=0.17 e A˚ 3
Table 1
Selected geometric parameters (A˚ ,).
O1—N1 1.225 (3)
O2—N1 1.219 (3)
N1—C1 1.464 (4)
N2—C8 1.339 (3)
N2—C9 1.340 (3)
C8—C9i 1.402 (3) C9—C8i
1.402 (3)
C4—O3—H3 109.5
O2—N1—O1 122.7 (3) O2—N1—C1 118.8 (3)
O1—N1—C1 118.5 (3) C8—N2—C9 120.3 (2)
Symmetry code: (i) 1x;y;1z.
Table 2
Hydrogen-bonding geometry (A˚ ,).
D—H A D—H H A D A D—H A
O3—H3 N2 0.82 2.01 2.818 (3) 170 C6—H6 O1ii
0.93 2.57 3.335 (4) 140 C2—H2 O2iii
0.93 2.52 3.380 (4) 154
Symmetry codes: (ii)x;2y;z; (iii) 2x;1y;z.
All H atoms were positioned geometrically and allowed to ride on their parent atoms, with Csp2—H = 0.93 A˚ with Uiso = 1.2Ueq(C), Csp3—H = 0.96 A˚ with U
iso= 1.5Ueq(C) and O—H = 0.82 A˚ with
Uiso= 1.2Ueq(O).
Data collection:SMART(Bruker, 2002); cell refinement:SAINT
(Bruker, 2002); data reduction:SAINT; program(s) used to solve structure:SHELXS97(Sheldrick, 1997); program(s) used to refine structure: SHELXL97 (Sheldrick, 1997); molecular graphics: XP
(Bruker, 2002); software used to prepare material for publication:XP
inSHELXTL(Bruker, 2002).
This work was supported by the Wenzhou Technology Project Foundation of China (No. S2004A004), the Zhejiang
Provincial Natural Science Foundation of China (No.
Y404118) and the National Natural Science Foundation of China (No. 20471043).
References
Bruker (2002).SADABS (Version 2.03), SAINT (Version 6.02), SMART
(Version 5.62) andSHELXTL(Version 6.10). Bruker AXS Inc., Madison, Wisconsin, USA.
Dong, W., Li, G., Shi, Z., Fu, W. & Zhang, D. (2003).Inorg. Chem. Commun.6, 776–780.
Kam, T. L., Michael, M., Hung, L. & Jack, T. T. (2005).FEMS Microbiol. Ecol.
51, 237–245.
Ng, S. W., Hu, S. Z., Hanna, J. V., Ral, S. S. S., Fun, H. K., Razak, I. A., Wojciechowski, G. & Brzezinski, B. (2001).J. Mol. Struct.595, 189–194. Paternostre, L., Damman, P. & Dosiere, M. (1998).Polymer,39, 4579–4592. Peng, W., Xin, J., Meiling, Q. & Lin, F.(2004).J. Chromatogr. B,813, 263–268. Sheldrick, G. M. (1997). SHELXS97 and SHELXL97. Release 97-2.
University of Go¨ttingen, Germany.
Smyth, M. V., Bailey, R. D. & Pennington, W. T. (1996).Acta Cryst.C52, 2170– 2173.
Su, L. L., Tsung, K. K., Wen, Y. C., Yu, S. L., Shih, Y. C., Shue, L. R., Ching, W. W., Hsi, C. L.& Chun, J. C. (2004).Neurosci. Lett.372, 40–45.
Tian, Y. & Yang, P. (1993).Chin. J. Inorg. Chem.9, 438–439.
Figure 2
supporting information
sup-1 Acta Cryst. (2005). E61, o614–o615
supporting information
Acta Cryst. (2005). E61, o614–o615 [https://doi.org/10.1107/S160053680500382X]
2,3,5,6-Tetramethylpyrazine
–
p
-nitrophenol (1/2)
Xiao-Qing Cai, Mao-Lin Hu, Ya-Juan Zhao, Ya-Qian Cheng and Shun Wang
2,3,5,6-Tetramethylpyrazine–p-nitrophenol (1/2)
Crystal data
C8H12N2·2C6H5NO3 Mr = 414.42 Triclinic, P1 Hall symbol: -P1
a = 4.7521 (2) Å
b = 8.8700 (4) Å
c = 13.1941 (6) Å
α = 73.916 (2)°
β = 81.771 (2)°
γ = 74.706 (2)°
V = 513.98 (4) Å3
Z = 1
F(000) = 218
Dx = 1.339 Mg m−3
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 1276 reflections
θ = 2.5–25.2°
µ = 0.10 mm−1 T = 298 K Block, colorless 0.48 × 0.35 × 0.15 mm
Data collection
Bruker APEX area-detector diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
φ and ω scans
Absorption correction: integration (SADABS; Bruker, 2002)
Tmin = 0.953, Tmax = 0.985
2747 measured reflections 1825 independent reflections 1720 reflections with I > 2σ(I)
Rint = 0.012
θmax = 25.2°, θmin = 1.6° h = −5→5
k = −8→10
l = −15→15
Refinement
Refinement on F2
Least-squares matrix: full
R[F2 > 2σ(F2)] = 0.077 wR(F2) = 0.170 S = 1.27 1825 reflections 139 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H-atom parameters constrained
w = 1/[σ2(F
o2) + (0.0532P)2 + 0.2196P]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max < 0.001
Δρmax = 0.16 e Å−3
Δρmin = −0.17 e Å−3
Special details
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2,
conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used
only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2
are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
O1 0.4149 (6) 0.9189 (3) −0.0822 (2) 0.1023 (10) O2 0.8091 (6) 0.7312 (3) −0.0677 (2) 0.0962 (9) O3 0.0826 (5) 0.4700 (3) 0.34784 (17) 0.0715 (7)
H3 0.1788 0.3775 0.3689 0.107*
N1 0.5647 (6) 0.7910 (3) −0.0341 (2) 0.0680 (7) N2 0.3737 (5) 0.1525 (2) 0.44480 (17) 0.0481 (6) C1 0.4426 (6) 0.7057 (3) 0.0671 (2) 0.0531 (7) C2 0.5856 (7) 0.5500 (4) 0.1141 (2) 0.0607 (8)
H2 0.7600 0.5004 0.0824 0.073*
C3 0.4678 (6) 0.4691 (3) 0.2080 (2) 0.0586 (8)
H3A 0.5626 0.3639 0.2399 0.070*
C4 0.2110 (6) 0.5423 (3) 0.2553 (2) 0.0510 (7) C5 0.0702 (7) 0.7000 (3) 0.2077 (2) 0.0604 (8)
H5 −0.1021 0.7507 0.2399 0.073*
C6 0.1863 (7) 0.7813 (3) 0.1126 (2) 0.0620 (8)
H6 0.0917 0.8861 0.0800 0.074*
C7 0.1485 (7) 0.0562 (4) 0.3285 (2) 0.0688 (9)
H7A 0.0617 0.1697 0.3064 0.103*
H7B −0.0028 −0.0020 0.3507 0.103*
H7C 0.2689 0.0212 0.2705 0.103*
C8 0.3327 (6) 0.0245 (3) 0.41888 (19) 0.0458 (6) C9 0.5373 (6) 0.1317 (3) 0.5245 (2) 0.0467 (7) C10 0.5772 (8) 0.2803 (4) 0.5484 (3) 0.0713 (9)
H10A 0.7619 0.3013 0.5161 0.107*
H10B 0.5731 0.2640 0.6236 0.107*
H10C 0.4222 0.3707 0.5209 0.107*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
supporting information
sup-3 Acta Cryst. (2005). E61, o614–o615
C6 0.071 (2) 0.0405 (15) 0.0606 (18) −0.0003 (14) −0.0101 (15) 0.0007 (13) C7 0.077 (2) 0.067 (2) 0.0560 (18) −0.0132 (17) −0.0106 (15) −0.0053 (15) C8 0.0488 (15) 0.0403 (14) 0.0432 (14) −0.0114 (11) 0.0040 (11) −0.0051 (11) C9 0.0519 (15) 0.0356 (13) 0.0474 (14) −0.0111 (11) 0.0063 (12) −0.0060 (11) C10 0.087 (2) 0.0457 (17) 0.083 (2) −0.0184 (16) −0.0055 (18) −0.0177 (16)
Geometric parameters (Å, º)
O1—N1 1.225 (3) C5—C6 1.381 (4)
O2—N1 1.219 (3) C5—H5 0.9300
O3—C4 1.354 (3) C6—H6 0.9300
O3—H3 0.8200 C7—C8 1.500 (4)
N1—C1 1.464 (4) C7—H7A 0.9600
N2—C8 1.339 (3) C7—H7B 0.9600
N2—C9 1.340 (3) C7—H7C 0.9600
C1—C6 1.370 (4) C8—C9i 1.402 (3)
C1—C2 1.380 (4) C9—C8i 1.402 (3)
C2—C3 1.373 (4) C9—C10 1.502 (4)
C2—H2 0.9300 C10—H10A 0.9600
C3—C4 1.374 (4) C10—H10B 0.9600
C3—H3A 0.9300 C10—H10C 0.9600
C4—C5 1.393 (4)
C4—O3—H3 109.5 C1—C6—H6 120.4
O2—N1—O1 122.7 (3) C5—C6—H6 120.4
O2—N1—C1 118.8 (3) C8—C7—H7A 109.5
O1—N1—C1 118.5 (3) C8—C7—H7B 109.5
C8—N2—C9 120.3 (2) H7A—C7—H7B 109.5
C6—C1—C2 121.3 (3) C8—C7—H7C 109.5
C6—C1—N1 119.3 (3) H7A—C7—H7C 109.5
C2—C1—N1 119.4 (3) H7B—C7—H7C 109.5
C3—C2—C1 119.3 (3) N2—C8—C9i 119.6 (2)
C3—C2—H2 120.3 N2—C8—C7 117.6 (2)
C1—C2—H2 120.3 C9i—C8—C7 122.8 (3)
C2—C3—C4 120.5 (3) N2—C9—C8i 120.1 (2)
C2—C3—H3A 119.7 N2—C9—C10 117.5 (2)
C4—C3—H3A 119.7 C8i—C9—C10 122.4 (3)
O3—C4—C3 123.7 (2) C9—C10—H10A 109.5
O3—C4—C5 116.6 (3) C9—C10—H10B 109.5
C3—C4—C5 119.7 (3) H10A—C10—H10B 109.5
C6—C5—C4 120.0 (3) C9—C10—H10C 109.5
C6—C5—H5 120.0 H10A—C10—H10C 109.5
C4—C5—H5 120.0 H10B—C10—H10C 109.5
C1—C6—C5 119.2 (3)
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
O3—H3···N2 0.82 2.01 2.818 (3) 170
C6—H6···O1ii 0.93 2.57 3.335 (4) 140
C2—H2···O2iii 0.93 2.52 3.380 (4) 154