metal-organic papers
Acta Cryst.(2007). E63, m793–m794 doi:10.1107/S1600536807006460 Renet al. [Li
2(C12H21N2Si)2]
m793
Acta Crystallographica Section E
Structure Reports Online
ISSN 1600-5368
Bis{
l
-
N
-[(
N
,
N
-dimethylamino)dimethylsilyl]-2,6-dimethylanilido}lithium(I)
Guang-Ming Ren,aDong-Lin
Shangband Jian-Ping Guob*
a
Department of Chemistry, Xinzhou Teachers’ University, Xinzhou 034000, People’s Republic of China, andbInstitute of Modern Chemistry,
Shanxi University, Taiyuan 030006, People’s Republic of China
Correspondence e-mail: guojp@sxu.edu.cn
Key indicators
Single-crystal X-ray study T= 203 K
Mean(C–C) = 0.005 A˚ Rfactor = 0.081 wRfactor = 0.170
Data-to-parameter ratio = 17.0
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
Received 31 January 2007 Accepted 6 February 2007
#2007 International Union of Crystallography All rights reserved
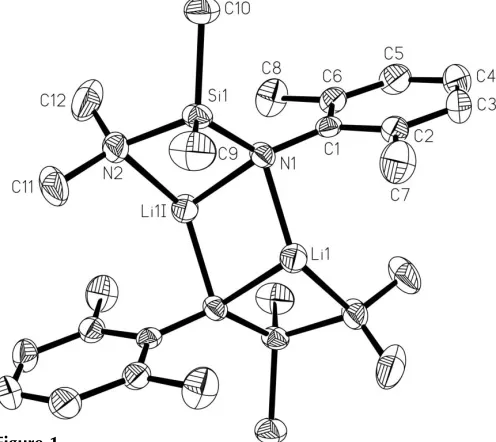
The dimeric lithium amide, [Li2(C12H21N2Si)2], exhibits a
four-rung ladder core of Li—N and Si—N bonds.
Comment
Lithium amides are useful reagents in syntheses (Lappert et al., 1980). The structural studies of these compounds could provide important insights in the prediction of properties and reactivities of the corresponding amido ligands. They have a high tendency to associate in the absence of neutral donor. Those with a low degree of aggregation can only be prepared in the presence of sterically very demanding substituents or strong donor molecules.
TheN-silylated class of anilides can be synthesized to have the desired steric and electronic properties, as exemplified by tetranuclear [LiN(SiMe3)(C6H5)]4, (I) (Antolini et al., 2000;
Bezombes et al., 2001). Its terminal Li atom makes an 6
contact with the phenyl ring (2.12 A˚ between Li and the centroid of the phenyl substituent); this distance is close to the length of the Li—N bond itself (2.0 A˚ ). The analogous compound [LiN(SiMe3)(2,6-iPr2C6H3)]2, (II), was the first
dimeric solvent-free lithium amide (Kennepohl et al., 1991), whose isopropyl substituents stabilize the dimeric structure of the compound. Following such studies on d–p interaction between Si and N atoms, we have incorporated an amino –NR2
(R= Me) pendant group, in the expectation that the pendant unit will bind to the Li. The title lithium anilide, [LiN(Si-Me2NMe2)(2,6-Me2C6H3)]2, (III), is another example of a
solvent-free dinuclear compound (Fig. 1).
Treatment of [NH(SiMe2NMe2)(2,6-Me2C6H3)] with Li
n
Bu in hexane gave compound (III). Reaction in diethyl ether did not give the solvent-coordinated species, as is the case with [(Et2O)Li{N(SiMe3)(2-C5H3N-6-Me)}]2 (Engelhardt et al.,
1988) and [(Et2O)Li{N(SiMe3)(C6H5)}]2(Antoliniet al., 2000;
Bezombes et al., 2001). Dimeric aggregation furnishes an (LiN)2rhombus with 2.0 A˚ sides and with 73.9 (3)angles. It
than those in [LiN(SiMe3)(C6H5)]4 [2.601 (9) A˚ ] and
[(Et2O)Li{N(SiMe3)(2-C5H3N-6-Me)}]2 [2.53 (1) A˚ ]. The Li
atom is coordinated by the the pendant amino group, so that the three-coordinate environment approximates a trigonal pyramid.
The molecule of (III) possesses a tricyclic ladder core. Such a feature has been observed in only a few examples, such as [LiN(SiMe2OMe){C(Ad) C(H)SiMe3}]2 (Ad is adamantyl)
and octanuclear [(hmqLi)8(THF)4] (hmq
is the 2-hydroxy-4-methylquinoline monoanion) (Antoliniet al., 2002; Liddle & Clegg, 2002).
Experimental
To a solution of [NH(SiMe2NMe2)(2,6-Me2C6H3)] (2.00 g,
9.00 mmol) in hexane (30 ml) was added a solution of LinBu (2.8M, 3.2 ml, 9.00 mmol) in hexane at 273 K. The reaction mixture was stirred at room temperature for a further 2 h. The resulting solution was concentrated to give the title compound as colourless crystals (yield 1.62 g, 79%). CHN analysis, calculated for C24H42Li2N4Si2:
C 63.12, H 9.27, N 12.27%; found: C 62.85, H 8.97, N 12.12%. Spectroscopic analysis:1H NMR (300 MHz, C
6D6,, p.p.m.): 7.22 (d,J
= 6.9 Hz, 2H, 3,5-H of phenyl), 6.86 (t,J= 7.4 Hz, 1H, 4-H of phenyl), 2.28 (s, 6H, NMe2), 2.10 (s, 6H, 2,6-Me2of phenyl), 0.13 (s, 6H,
SiMe2); 13
CNMR (75 MHz, C6D6,, p.p.m.): 155.2 (1-C of phenyl),
134.0 (2,6-C of phenyl), 131.3 (3,5-C of phenyl), 120.3 (4-C of phenyl), 41.2 (NMe2), 23.5 (2,6-Me2of phenyl), 2.5 (SiMe2)
Crystal data
[Li2(C12H21N2Si)2]
Mr= 456.68
Monoclinic, P21=n
a= 9.7958 (9) A˚
b= 10.5049 (11) A˚
c= 14.0597 (14) A˚
V= 1400.7 (2) A˚3
Z= 2
MoKradiation = 0.14 mm1
T= 203 (2) K 0.300.300.20 mm
Data collection
Bruker SMART CCD area-detector diffractometer
Absorption correction: multi-scan (SADABS; Sheldrick, 1996)
Tmin= 0.700,Tmax= 0.972
5654 measured reflections 2461 independent reflections 2305 reflections withI> 2(I)
Rint= 0.028
Refinement
R[F2> 2(F2)] = 0.081
wR(F2) = 0.170
S= 1.28 2461 reflections
145 parameters
H-atom parameters constrained max= 0.30 e A˚3
min=0.20 e A˚3
Table 1
Selected geometric parameters (A˚ ,).
Si1—N1 1.686 (3)
Si1—N2 1.767 (3)
N1—C1 1.402 (4)
N1—Li1 1.980 (6)
Li1—N1i
2.006 (6) Li1—N2i
2.063 (6)
N1—Si1—N2 102.12 (13)
C1—N1—Li1 96.0 (2)
Si1—N1—Li1 126.3 (2)
C1—N1—Li1i 133.9 (3) N1—Li1—N1i 106.1 (3) N1—Li1—N2i 135.1 (3)
Symmetry code: (i)xþ1;yþ2;z.
All H atoms were initially located in a difference Fourier map. The methyl H atoms were then constrained to an ideal geometry, with C— H = 0.98 A˚ andUiso(H) = 1.5Ueq(C), but each group was allowed to
rotate freely about its C—C bond. The other H atoms were placed in geometrically idealized positions and constrained to ride on their parent atoms, with C—H distances in the range 0.95–1.00 A˚ and
Uiso(H) = 1.2Ueq(C).
Data collection:SMART(Bruker, 2000); cell refinement:SAINT
(Bruker, 2000); data reduction:SAINT; program(s) used to solve structure:SHELXS97(Sheldrick, 1997); program(s) used to refine structure: SHELXL97 (Sheldrick, 1997); molecular graphics:
SHELXTL/PC(Sheldrick, 1999); software used to prepare material for publication:SHELXTL/PC.
This work was carried out under the sponsorship of the National Natural Science Foundation of China (grant No. 50443001).
References
Antolini, F., Gehrhus, B., Hitchcock, P. B. & Lappert, M. F. (2002).Angew. Chem. Int. Ed.41, 2568–2571.
Antolini, F., Hitchcock, P. B., Lappert, M. F. & Merle, P. G. (2000).Chem. Commun.pp. 1301–1302.
Bezombes, J. P., Hitchcock, P. B., Lappert, M. F. & Merle, P. G. (2001).J. Chem. Soc. Dalton Trans.pp. 816–821.
Bruker (2000).SMART(Version 5.0) andSAINT(Version 6.02). Bruker AXS Inc., Madison, Wisconsin, USA.
Engelhardt, L. M., Jacobsen, G. E., Junk, P. C., Raston, C. L. & Skelton, B. W. (1988).J. Chem. Soc. Dalton Trans.pp. 1011–1020.
Kennepohl, D. K., Brooker, S., Sheldrick, G. M. & Roesky, H. W. (1991).
Chem. Ber.124, 2223–2225.
Lappert, M. F., Power, P. P., Sanger, A. R. & Srivastava, R. C. (1980).Metal and Metalloid Amides, pp. 24–44. Chichester: Ellis Horwood.
Liddle, S. T. & Clegg, W. (2002). J. Chem. Soc. Dalton Trans.pp. 3923– 3924.
Sheldrick, G. M. (1996).SADABS. University of Go¨ttingen, Germany. Sheldrick, G. M. (1997). SHELXL97 and SHELXS97. University of
Go¨ttingen, Germany.
[image:2.610.48.296.74.295.2]Sheldrick, G. M. (1999).SHELXTL/PC. Version 6.10. Bruker AXS Inc., Madison, Wisconsin, USA.
Figure 1
supporting information
sup-1
Acta Cryst. (2007). E63, m793–m794
supporting information
Acta Cryst. (2007). E63, m793–m794 [https://doi.org/10.1107/S1600536807006460]
Bis{
µ
-
N
-[(
N
,
N
-dimethylamino)dimethylsilyl]-2,6-dimethylanilido}lithium(I)
Guang-Ming Ren, Dong-Lin Shang and Jian-Ping Guo
Bis{µ-N-[(N,N-dimethylamino)dimethylsilyl]-2,6-dimethylanilido}lithium(I)
Crystal data
[Li2(C12H21N2Si)2]
Mr = 456.68
Monoclinic, P21/n
a = 9.7958 (9) Å
b = 10.5049 (11) Å
c = 14.0597 (14) Å
β = 104.499 (2)°
V = 1400.7 (2) Å3
Z = 2
F(000) = 496
Dx = 1.083 Mg m−3
Melting point = 173–174 K Mo Kα radiation, λ = 0.71073 Å Cell parameters from 2419 reflections
θ = 2.5–27.6°
µ = 0.14 mm−1
T = 203 K Prism, colourless 0.30 × 0.30 × 0.20 mm
Data collection
Bruker SMART CCD area-detector diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
ω scans
Absorption correction: multi-scan (SADABS; Sheldrick, 1996)
Tmin = 0.700, Tmax = 0.972
5654 measured reflections 2461 independent reflections 2305 reflections with I > 2σ(I)
Rint = 0.028
θmax = 25.0°, θmin = 2.3°
h = −11→9
k = −9→12
l = −16→16
Refinement
Refinement on F2
Least-squares matrix: full
R[F2 > 2σ(F2)] = 0.081
wR(F2) = 0.170
S = 1.28 2461 reflections 145 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H-atom parameters constrained
w = 1/[σ2(F
o2) + (0.0494P)2 + 1.123P]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max < 0.001
Δρmax = 0.30 e Å−3
Δρmin = −0.20 e Å−3
Special details
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2,
conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used
only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2
are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
supporting information
sup-3
Acta Cryst. (2007). E63, m793–m794
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
Si1 0.0439 (5) 0.0441 (5) 0.0475 (5) 0.0068 (4) 0.0185 (4) 0.0143 (4) N1 0.0440 (15) 0.0326 (14) 0.0449 (15) 0.0038 (11) 0.0142 (12) 0.0022 (12) N2 0.094 (2) 0.0501 (18) 0.0548 (19) 0.0175 (17) 0.0428 (18) 0.0100 (15) C1 0.0415 (17) 0.0403 (17) 0.0310 (15) 0.0047 (14) 0.0098 (13) 0.0095 (13) C2 0.058 (2) 0.0409 (19) 0.0429 (18) 0.0008 (16) 0.0116 (16) 0.0010 (15) C3 0.091 (3) 0.053 (2) 0.056 (2) 0.010 (2) 0.023 (2) −0.0117 (19) C4 0.081 (3) 0.087 (3) 0.065 (3) 0.038 (3) 0.029 (2) −0.004 (2) C5 0.047 (2) 0.095 (3) 0.058 (2) 0.017 (2) 0.0156 (18) 0.005 (2) C6 0.0397 (18) 0.064 (2) 0.0407 (18) 0.0060 (16) 0.0103 (15) 0.0017 (16) C7 0.076 (3) 0.070 (3) 0.078 (3) −0.022 (2) 0.014 (2) −0.020 (2) C8 0.051 (2) 0.097 (3) 0.072 (3) −0.019 (2) 0.014 (2) −0.020 (3) C9 0.050 (2) 0.100 (4) 0.098 (3) 0.002 (2) 0.034 (2) 0.028 (3) C10 0.073 (3) 0.059 (2) 0.057 (2) 0.010 (2) 0.018 (2) 0.0177 (19) C11 0.163 (5) 0.077 (3) 0.105 (4) 0.057 (3) 0.104 (4) 0.035 (3) C12 0.160 (5) 0.072 (3) 0.060 (3) −0.019 (3) 0.046 (3) −0.016 (2) Li1 0.063 (4) 0.041 (3) 0.047 (3) −0.002 (3) 0.019 (3) 0.000 (3)
Geometric parameters (Å, º)
Si1—N1 1.686 (3) C7—H7B 0.9600 Si1—N2 1.767 (3) C7—H7C 0.9600 Si1—C10 1.861 (4) C8—H8A 0.9600 Si1—C9 1.864 (4) C8—H8B 0.9600 Si1—Li1i 2.602 (6) C8—H8C 0.9600
N1—C1 1.402 (4) C9—H9A 0.9600 N1—Li1 1.980 (6) C9—H9B 0.9600 N1—Li1i 2.006 (6) C9—H9C 0.9600
N2—C12 1.460 (6) C10—H10A 0.9600 N2—C11 1.469 (5) C10—H10B 0.9600 N2—Li1i 2.063 (6) C10—H10C 0.9600
C1—C2 1.402 (4) C11—H11A 0.9600 C1—C6 1.410 (4) C11—H11B 0.9600 C1—Li1 2.542 (6) C11—H11C 0.9600 C2—C3 1.380 (5) C12—Li1i 2.740 (7)
C2—C7 1.507 (5) C12—H12A 0.9600 C3—C4 1.359 (6) C12—H12B 0.9600 C3—H3A 0.9300 C12—H12C 0.9600 C4—C5 1.365 (6) Li1—N1i 2.006 (6)
C4—H4A 0.9300 Li1—N2i 2.063 (6)
C5—C6 1.379 (5) Li1—Li1i 2.397 (11)
C5—H5A 0.9300 Li1—Si1i 2.602 (6)
C6—C8 1.498 (5) Li1—C12i 2.740 (7)
C7—H7A 0.9600
N1—Si1—C10 113.81 (16) H8A—C8—H8C 109.5 N2—Si1—C10 111.60 (17) H8B—C8—H8C 109.5 N1—Si1—C9 116.19 (18) Si1—C9—H9A 109.5 N2—Si1—C9 106.2 (2) Si1—C9—H9B 109.5 C10—Si1—C9 106.67 (19) H9A—C9—H9B 109.5 N1—Si1—Li1i 50.43 (15) Si1—C9—H9C 109.5
N2—Si1—Li1i 52.19 (15) H9A—C9—H9C 109.5
C10—Si1—Li1i 133.89 (19) H9B—C9—H9C 109.5
C9—Si1—Li1i 119.1 (2) Si1—C10—H10A 109.5
C1—N1—Si1 128.9 (2) Si1—C10—H10B 109.5 C1—N1—Li1 96.0 (2) H10A—C10—H10B 109.5 Si1—N1—Li1 126.3 (2) Si1—C10—H10C 109.5 C1—N1—Li1i 133.9 (3) H10A—C10—H10C 109.5
Si1—N1—Li1i 89.2 (2) H10B—C10—H10C 109.5
Li1—N1—Li1i 73.9 (3) N2—C11—H11A 109.5
C12—N2—C11 111.0 (4) N2—C11—H11B 109.5 C12—N2—Si1 116.7 (3) H11A—C11—H11B 109.5 C11—N2—Si1 121.2 (3) N2—C11—H11C 109.5 C12—N2—Li1i 100.8 (3) H11A—C11—H11C 109.5
C11—N2—Li1i 117.7 (3) H11B—C11—H11C 109.5
Si1—N2—Li1i 85.2 (2) N2—C12—Li1i 47.7 (2)
N1—C1—C2 122.4 (3) N2—C12—H12A 109.5 N1—C1—C6 120.2 (3) Li1i—C12—H12A 87.2
C2—C1—C6 117.2 (3) N2—C12—H12B 109.5 N1—C1—Li1 50.77 (18) Li1i—C12—H12B 78.8
C2—C1—Li1 94.4 (2) H12A—C12—H12B 109.5 C6—C1—Li1 122.1 (3) N2—C12—H12C 109.5 C3—C2—C1 120.5 (3) Li1i—C12—H12C 156.4
C3—C2—C7 119.1 (3) H12A—C12—H12C 109.5 C1—C2—C7 120.4 (3) H12B—C12—H12C 109.5 C4—C3—C2 121.7 (4) N1—Li1—N1i 106.1 (3)
C4—C3—H3A 119.2 N1—Li1—N2i 135.1 (3)
C2—C3—H3A 119.2 N1i—Li1—N2i 82.6 (2)
C3—C4—C5 118.8 (4) N1—Li1—Li1i 53.5 (2)
C3—C4—H4A 120.6 N1i—Li1—Li1i 52.5 (2)
C5—C4—H4A 120.6 N2i—Li1—Li1i 118.5 (4)
C4—C5—C6 121.9 (4) N1—Li1—C1 33.28 (13) C4—C5—H5A 119.1 N1i—Li1—C1 126.7 (3)
C6—C5—H5A 119.1 N2i—Li1—C1 106.7 (3)
C5—C6—C1 120.0 (3) Li1i—Li1—C1 79.0 (3)
C5—C6—C8 119.3 (3) N1—Li1—Si1i 126.6 (3)
C1—C6—C8 120.7 (3) N1i—Li1—Si1i 40.39 (13)
C2—C7—H7A 109.5 N2i—Li1—Si1i 42.58 (14)
C2—C7—H7B 109.5 Li1i—Li1—Si1i 81.7 (3)
H7A—C7—H7B 109.5 C1—Li1—Si1i 122.4 (2)
C2—C7—H7C 109.5 N1—Li1—C12i 154.8 (3)
H7A—C7—H7C 109.5 N1i—Li1—C12i 93.9 (2)
supporting information
sup-5
Acta Cryst. (2007). E63, m793–m794
C6—C8—H8A 109.5 Li1i—Li1—C12i 143.5 (4)
C6—C8—H8B 109.5 C1—Li1—C12i 121.6 (3)
H8A—C8—H8B 109.5 Si1i—Li1—C12i 61.94 (16)